SUBSTITUIERTE N-ARYLETHYL-2-ARYLCHINOLIN-4-CARBOXAMIDE
UND IHRE VERWENDUNG
Die vorliegende Anmeldung betrifft neue substituierte /V-Arylethyl-2-arylchinolin-4-carboxamid- Derivate, Verfahren zu ihrer Herstellung, ihre Verwendung allein oder in Kombinationen zur Be- handlung und/oder Prävention von Krankheiten sowie ihre Verwendung zur Herstellung von Arzneimitteln zur Behandlung und/oder Prävention von Krankheiten, insbesondere zur Behandlung und/oder Prävention von fibrotischen und inflammatorischen Erkrankungen.
Prostaglandin F2alpha (PGF2a) gehört zur Familie der bioaktiven Prostaglandine, die Derivate der Arachidonsäure darstellen. Nach Freisetzung aus Membranphospholipiden durch A2-Phos- pholipasen wird die Arachidonsäure durch Cyclooxygenasen zu dem Prostaglandin H2 (PGH2) oxidiert, welches durch die PGF-Synthase weiter zu PGF2a umgewandelt wird. Zu einem wesentlich geringeren Anteil kann PGF2a auch aus anderen Prostaglandinen wie PGE2 oder PGD2 enzymatisch gebildet werden [Watanabe et al., J. Biol. Chem. 1985, 260,7035-7041]. PGF2a wird nicht gespeichert, sondern nach der Synthese sofort freigesetzt, wodurch es lokal seine Wirkungen entfaltet. PGF2a ist ein instabiles Molekül ( /2 < 1 Minute), welches schnell enzymatisch in Lunge, Leber und Niere zu einem inaktiven Metaboliten, 15-Ketodihydro-PGF2a, umgelagert wird [Basu et al., Acta Chem. Scand. 1992, 46, 108-1 10]. 15-Ketodihydro-PGF2a ist in größeren Mengen im Plasma und später auch im Urin sowohl unter physiologischen als auch pathophysiologischen Bedingungen detektierbar.
Die biologischen Effekte von PGF2a kommen durch die Bindung und Aktivierung eines membranständigen Rezeptors, des PGF2a-Rezeptors oder auch so genannten FP-Rezeptors, zustande. Der FP-Rezeptor gehört zu den G-Protein-gekoppelten Rezeptoren, die durch sieben Transmembrandomänen charakterisiert sind. Neben dem humanen FP-Rezeptor konnten auch die FP-Rezeptoren von Maus und Ratte kloniert werden [Abramovitz et al., J. Biol. Chem. 1994, 269, 2632-2636; Sugimoto et al., J. Biol. Chem. 1994, 269, 1356-1360; Kitanaka et al., Prostaglandins 1994, 48, 31-41]. Im Menschen existieren zwei Isoformen des FP-Rezeptors, FPA und FPB. Von den Prostanoid-Rezeptoren ist der FP-Rezeptor am wenigsten selektiv, da an ihn neben PGF2a noch PGD2 und PGE2 mit nanomolaren Affinitäten binden [Woodward et al., Phar- macol. Rev. 2011 , 63, 471-538]. Stimulation des FP-Rezeptors führt primär zur Gq-abhängigen Aktivierung der Phospholipase C, was in Freisetzung von Calcium und einer Aktivierung der Di- acylglycerol-abhängigen Proteinkinase C (PKC) resultiert. Der erhöhte intrazelluläre Calcium- spiegel führt zur Calmodulin-vermittelten Stimulation der Myosin-Ieichte Ketten-Kinase (MLCK). Neben der Kopplung an das G-Protein Gq kann der FP-Rezeptor über G12/G13 auch die Rho/Rhokinase-Signaltransduktionskaskade aktivieren sowie über Gi-Kopplung alternativ den Raf/MEK/MAP-Signalweg stimulieren [Woodward et al., Pharmacol. Rev. 2011 , 63, 471-538].
PGF2a ist an der Regulation zahlreicher physiologischer Funktionen, wie z.B. Ovarialfunktionen, Embryonalentwicklung, Veränderungen in der Gebärmutterschleimhaut, Gebärmutterkontraktion, Luteolyse und in der Induktion von Geburtswehen und der Entbindung, beteiligt. PGF2a wird auch im Endometrium in Epithelialzellen synthetisiert, wo es die zelluläre Proliferation stimuliert [Woodward et al., Pharmacol. Rev. 2011 , 63, 471-538]. Außerdem ist PGF2a ein potenter Sti- mulator der Glattmuskel-, Gefäß- und Bronchokonstriktion und ist in akuten und chronischen inflammatorischen Prozessen involviert [Basu, Mol. Cells 2010, 30, 383-391]. So konnte gezeigt werden, dass 15-keto-dihydro-PGF2a, ein stabiler Metabolit von PGF2a, systemisch in Patienten mit rheumathoider Arthrose, psoriatischer Arthrose und Osteoarthrose, detektiert werden konnte. In der Niere ist PGF2a an der Wasserabsorption, Natriurese und Diurese beteiligt. In den Augen reguliert PGF2a den intraokularen Druck. PGF2a spielt auch eine wichtige Rolle im Knochenmetabolismus: Das Prostaglandin stimuliert den Natrium-abhängigen Transport von anorganischem Phosphat in Osteoblasten und es steigert die Freisetzung von lnterleukin-6 und des vaskulären endothelialen Wachstumsfaktors (VEGF) in Osteoblasten; außerdem ist PGF2a ein starkes Mi- togen und ein Überlebensfaktor für Osteoblasten [Agas et al., J. Cell P ysiol. 2013, 228, 25-29].
Erhöhte PGF2a / FP Rezeptor Aktivität führte auch zur Hochregulation von tumorigenen und an- giogenen Genen wie COX-2 [Sales et al., 2007, Endocrinology 148:3635-44], FGF-2 und VEGF [Sales et al., 2010 Am J Pathol 176:431] was darauf hinweist, dass der FP Rezeptor endometria- les Tumorwachstum durch Regulation von vaskulären Funktionen stimuliert. Darüber hinaus ist der FP Rezeptor an der Regulation der Proliferation endometrialer Epithelialzellen beteiligt und kann deren Adhäsion an die extrazelluläre Matrix und Motilität beeinflussen. Die Befunde deuten darauf hin, dass PGF2a / FP Receptor eine multifaktorielle Rolle in endometrialen Adenokarzinomen spielt [Yang et al., 2013 J Recept Signal Transduct, 33(1 ): 14-27/
Erhöhte Expression des FP Rezeptors in Vorläuferzellen von Oligodendrozyten (OPCs) könnte ein Marker für Schädigung von Oligodendrozyten und aktivem Myelin sein [Soldan et al., Neuro- logy 2015, 84]. Nach Autopsie konnte in Geweben von Patienten mit multipler Sklerose (MS) die Expression des FP Rezeptors auf OPCs in der Nähe der Ränder von MS Plaques beobachtet werden. Keine FP Rezeptorexpression wurde in Kontrollproben weißer Gehirnsubstanz gefunden. Dies deutet darauf hin, dass der FP Rezeptor eine Rolle in der Entstehung von multipler Sklerose spielt [Carlson et al., 2015, Mult. Sclerosis. 23 (1 1 ) 467-468].
Verletzungen des Gehirns führen zur Hochregulation von Prostaglandinen, vor allem des proinflammatorischen PGF2a und zur Überaktivierung des FP Rezeptors. So konnte mit FP Rezeptor defizienten Mäusen und Behandlung mit dem FP-Antagonisten AL-8810 signifikante neuropro- tektive Effekte nach Okklusion der Zerebralarterie gezeigt werden [Kim et al., 2012, Neurobiol Disease 48, 58-65].
Darüber hinaus konnte gezeigt werden, dass die PGF2a-FP-Rezeptoraktivierung in verschiedenen kardiovaskulären Dysfunktionen, wie z.B. Myokardialer Fibrose, Myokardinfarkt und Hyper- tension, involviert ist [Zhang et al., Frontiers in Pharmacol. 2010, 1, 1-7; Ding et al., Int. J. Bio- chem. Cell. Bio!., 2012, 44, 1031-1039; Ding et al., J. Mol. Med., 2014, 6, 629-640].
So ist der Hauptmetabolit von PGF2a, 15-keto-dihydro-PGF2a bei Menschen mit Lebensbedingungen mit erhöhtem kardiovaskulären Risiko [Helmersson-Karlquist et al., Eur Heart J 2015, 36, 238-243], wie z.B. auch bei Rauchern [Helmersson et al., 2005 Atherosclerosis 181 , 201- 207), Fettleibigkeit [Sinaiko et al., 2005 Circulation 1 1 1 , 1985-1991], Diabetes Typ I [Basu et al., 2005, 28, 1371 -1375] und Diabetes Typ II [Helmersson et al., 2004, Circulation 109, 1729-1734] erhöht. [Zhang et al., Frontiers in Pharmacol 2010, 1 :1-7]. Auch konnte gezeigt werden, dass ein genetischer Polymorphismus in einer chinesischen Subpopulation zu erhöhter Transkription des FP Genes und erhöhter Vasokontraktilität führt [Xiao et al., 2015, Arterioscler Thromb Vase Biol. 35:1687-1695].
Außerdem ist der PGF2a-Rezeptor (FP) bei Gelenksentzündungen und der Regulation der Sig- nalkaskade des knochenmorphogenetischen Proteins (BMP) beteiligt und fördert die Differenzierung von Chondrozyten [Kim et al., Biochim. Biophys. Acta, 2015, 1853, 500-512]. Stabilere Analoga von PGF2a wurden zur Östrussynchronisierung und zur Beeinflussung humaner Reproduktionsfunktionen entwickelt sowie zur Reduktion des intraokularen Drucks für die Behandlung des Glaukoms eingesetzt [Basu, Mol. Cells 2010, 30, 383-391]. Bei letzterer Anwendung wurde als Nebenwirkung die Stimulation von Haarwachstum, z.B. das von Wimpern, durch die chemisch stabileren PGF2a Analoga wie z.B. Latanoprost beobachtet. [Johnston et al., Am J Ophthalmol 1997, 124-544-547]. Auch werden die Gene des FP Rezeptors in menschlichen Haarfollikeln der Kopfhaut exprimiert [Khidhir et al., J Invest Dermatol, 2009, Abstr 607]. Diese Befunde legen die Vermutung nahe, dass der FP Rezeptor an der Regulation des Haarwachstums beteiligt ist und auch bei Erkrankungen wie z.B. Hirsutismus beteiligt sein kann.
Auch ist die Rolle des FP-Rezeptors in der Signalkaskade bei der Entstehung von viszeralem Schmerz (Dysmenorrhöe) gut beschrieben. So korreliert die dysmenorrhoischer Schmerz am besten mit der Rate der PGF-Freisetzung während der Menstruation (vgl. Powell et al., Prostaglandins 1985, 29, 273-290; Dawood and Khan-Dawood, Am. J. Obstet. Gynecol. 2007, 196, 35.e1-35.e5; Hsia et al., Endocrinology 2011 , 152, 2090-2099). Ein Zusammenhang des FP-Rezeptor- Signalwegs in periphär vermitteltem inflammatorischen Schmerz wurde bislang nicht beschrieben. Die hiermit vorgelegten Daten zeigen diesen überraschenden Zusammenhang erstmals.
In Patienten mit idiopathischer pulmonaler Fibrose (IPF) konnte gezeigt werden, dass der stabile PGF2a-Metabolit 15-ketodihydro-PGF2a im Plasma signifikant erhöht ist und dass die Spiegel von 15-ketodihydro-PGF2a mit funktionalen Parametern, wie z.B. der forcierten Vitalkapazität (FVC), der Diffusionsstrecke von Kohlenmonoxid in der Lunge (DLCO) und dem 6-Minuten-
Gehtest, korrelieren. Außerdem konnte ein Zusammenhang zwischen erhöhtem Plasma-15- ketodihydro-PGF2a und der Mortalität der Patienten festgestellt werden [Aihara et al., PLoS One 2013, 8, 1-6]. In Übereinstimmung damit wurde auch gezeigt, dass die Stimulation von humanen Lungenfibroblasten mit natürlich vorkommenden Silikastäuben, die im Menschen bei chronischer Inhalation zu Silikose und als Folge Lungenfibrose führen können, eine starke Hochregulation der PGF2a-Synthese bewirkt [O'Reilly et al., Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 288, L1010-L1016]. In der Bleomycin-induzierten Lungenfibrose in Mäusen führte das Ausschalten des FP-Rezeptors durch Knock-down (FP-/-) zu deutlich reduzierter pulmonaler Fibrose im Vergleich zu Wildtyp-Mäusen [Oga et al., Nat. Med. 2009, 15, 1426-1430]. In FP-/- Mäusen war nach Bleomycin-Gabe eine signifikante Reduktion des Hydroxyprolingehalts sowie ein verminderte Induktion von profibrotischen Genen im Lungengewebe zu sehen. Außerdem war die Funktion der Lunge in FP-/- Mäusen im Vergleich zu den Wildtyp-Mäusen deutlich verbessert. In humanen Lungenfibroblasten stimuliert PGF2a die Proliferation und die Kollagenproduktion über den FP-Rezeptor. Da dies unabhängig von dem profibrotischen Mediator TGFß erfolgt, stellt die PGF2a/ FP-Rezeptor-Signalkaskade einen eigenständigen Weg bei der Entstehung der Lungenfibrose dar [Oga et al., Nat. Med. 2009, 15, 1426-1430]. Diese Befunde zeigen, dass der FP- Rezeptor ein therapeutisches Zielprotein zur Behandlung von IPF ist [Olman, Nat. Med. 2009, 15, 1360-1361]. Die Beteiligung von PGF2a bei der Induktion von fibrotischen Veränderungen konnte auch an kardialen Maus-Fibroblasten [Ding et al., Int. J. Biochem. & Cell Biol. 2012, 44, 1031-1039], in einem Tiermodell der Sklerodermie [Kanno et al., Arthritis Rheum. 2013, 65, 492- 502] sowie an Synoviozyten aus Patienten mit Kniegelenksarthrose [Bastiaansen et al. Arthritis Rheum. 2013, 65, 2070-2080] gezeigt werden.
Es wird daher angenommen, dass der FP-Rezeptor bei vielen Erkrankungen, Verletzungen und pathologischen Veränderungen, deren Entstehung und/oder Progression mit einem entzündli- chen Geschehen und/oder einem proliferativen und fibroproliferativen Gewebe- und Gefäßumbau in Zusammenhang steht, eine wichtige Rolle spielt. Dies können insbesondere Erkrankungen und/ oder Schädigungen der Lunge, des Herz-Kreislauf-Systems oder der Niere sein, oder es kann sich hierbei um eine Erkrankung des Blutes, eine Krebs-Erkrankung oder um andere entzündliche Erkrankungen handeln.
In diesem Zusammenhang zu nennende inflammatorische und fibrotische Erkrankungen und Schädigungen der Lunge sind insbesondere die idiopathische Lungenfibrose, interstitielle Lungenerkrankungen assoziert mit rheumatoider Arthrose, die pulmonale Hypertonie, das Bronchiolitis obliterans-Syndrom (BOS), die chronisch-obstruktive Lungenerkrankung (COPD), Asthma und zystische Fibrose. Erkrankungen und Schädigungen des Herz-Kreislauf-Systems, in denen der FP-Rezeptor involviert ist, sind zum Beispiel Gewebeveränderungen nach einem Myokardinfarkt und bei der Herzinsuffizienz. Erkrankungen der Niere sind zum Beispiel Niereninsuffizienz
und Nierenversagen. Eine Erkrankung des Blutes ist zum Beispiel die Sichelzellanämie. Beispiele für einen Gewebeab- und -umbau bei Krebsprozessen sind das Einwandern von Krebszellen in das gesunde Gewebe (Metastasenbildung) und die Neuausbildung von versorgenden Blutgefäßen (Neo-Angiogenese). Andere entzündliche Krankheiten, bei denen der FP-Rezeptor eine Rolle spielt, sind zum Beispiel Arthrose und Multiple Sklerose.
Die idiopathische Lungenfibrose oder idiopathische pulmonale Fibrose (IPF) ist eine progrediente Lungenerkrankung, die unbehandelt durchschnittlich innerhalb von 2.5 bis 3.5 Jahren nach Diagnosestellung zum Tode führt. Die Patienten sind zum Zeitpunkt der Diagnosestellung meist älter als 60 Jahre, und Männer sind etwas häufiger betroffen als Frauen. Der Krankheitsbeginn der IPF ist schleichend und durch eine zunehmende Atemnot und trockenen Reizhusten gekennzeichnet. IPF gehört zur Gruppe der idiopathischen interstitiellen Pneumonien (IIP), einer heterogenen Gruppe von Lungenerkrankungen, die durch Fibrose und Inflammation unterschiedlichen Grades charakterisiert sind und die mit Hilfe klinischer, bildgebender und feingeweblicher Kriterien unterschieden werden. Innerhalb dieser Gruppe hat die idiopathische pulmonale Fibrose aufgrund ihrer Häufigkeit und des aggressiven Verlaufs eine besondere Bedeutung [Ley et al., Am. J. Respir. Crit. Care Med. 2011 , 183, 431-440]. Die IPF kann entweder sporadisch oder familiär gehäuft auftreten. Die Ursachen sind derzeit nicht geklärt. In den letzten Jahren wurden jedoch zahlreiche Hinweise dafür gefunden, dass eine chronische Schädigung des Alveolarepithels zur Freisetzung von pro- fibrotischen Zytokinen/Mediatoren führt, gefolgt von einer gesteigerten Fibroblastenproliferation und einer vermehrten Kollagenfaserbildung, wodurch es zu einer fleckenförmigen Fibrose und der typischen honigwabenartigen Struktur der Lunge kommt [Strieter et al.,Chest 2009, 136, 1364- 1370]. Die klinischen Folgen der Fibrosierung sind eine Abnahme der Elastizität des Lungengewebes, eine Verminderung der Diffusionskapazität sowie die Entwicklung einer schweren Hypoxie. Lungenfunktionell kann entsprechend eine Verschlechterung der forcierten Vitalkapazität (FVC) und der Diffusionskapazität (DLCO) festgestellt werden. Wesentliche und prognostisch bedeutende Komorbiditäten der IPF sind die akute Exazerbation und die pulmonale Hypertonie [von der Beck et al., Der Pneumologe 2013, 10(2), 105-11 1]. Die Prävalenz der pulmonalen Hypertonie bei interstitiellen Lungenerkrankungen liegt bei 10-40% [Lettieri et al, Chest 2006, 129, 746-752; Behr et al., Eur. Respir. J. 2008, 31, 1357-1367]. Es gibt gegenwärtig keine kurative Behandlung für die IPF - mit Ausnahme der Lungentransplantation.
Die Rheumatoide Arthritis (RA) ist eine fortschreitende, systemische Autoimmunerkrankung, welche durch eine chronische erosive Synovitis charakterisiert wird. Die interstitielle Lungenerkrankung (ILD) ist eine der häufigsten extraartikulären Manifestationen von RA [Wells et al. Nat Rev Rheumatol 2014, 10, 728-739]. Ungefähr 10% der Patienten mit RA haben eine klinisch nachgewiesene interstitielle Lungenerkrankung (RA-ILD), ein weiteres Drittel zeigt eine subklinische ILD bei CT-Aufnahmen der Brust. Die Sterblichkeitsrate für Patienten mit RA-ILD ist dreimal so hoch wie für Patienten mit RA ohne ILD, mit einer durchschnittlichen Lebenserwar-
tung von nur 2,6 Jahren nach ILD Diagnose [Olson et al. Am J Respir Crit Care Med 2011 183, 372-378; Doyle et al. Chest 2014, 145(3), 454-463].
Die inflammatorischen und autoimmunen Veränderungen der Lunge zusammen mit verschiedenen umweltbezogenen Auslösern (e.g. Rauchen, Feinstaub, chemische Reizmittel) und einer genetischen Disposition, spielen eine wichtige Rolle in der Entwicklung und Progression der RA- ILD [Catrina et al. Nat Rev Rheumatol 2014, 10(1 1 ), 645-653].
Die Pulmonale Hypertonie (PH) ist eine progrediente Lungenerkrankung, die unbehandelt durchschnittlich innerhalb von 2.8 Jahren nach Diagnosestellung zum Tode führt. Definitionsgemäß liegt bei einer chronischen pulmonalen Hypertonie ein pulmonal-arterieller Mitteldruck (mPAP) von > 25 mmHg in Ruhe oder > 30 mmHg unter Belastung vor (Normalwert < 20 mmHg). Die Patho- physiologie der pulmonalen Hypertonie ist gekennzeichnet durch Vasokonstriktion und ein Remodeling der Pulmonalgefäße. Bei der chronischen PH kommt es zu einer Neomuskularisierung primär nicht muskularisierter Lungengefäße, und die Gefäßmuskulatur der bereits muskularisierten Gefäße nimmt an Umfang zu. Durch diese zunehmende Obliteration der Lungenstrombahn kommt es zu einer progredienten Belastung des rechten Herzens, die zu einer verminderten Auswurfleistung des rechten Herzens führt und letztlich in einem Rechtsherzversagen endet [M. Humbert et al., J. Am. Coli. Cardio!. 2004, 43, 13S-24S]. Wenn auch die idiopathische (oder primäre) pul- monal-arterielle Hypertonie (IPAH) eine sehr seltene Erkrankung ist, so ist die sekundäre pulmonale Hypertonie (non-PAH PH, NPAHPH) weit verbreitet, und es wird zur Zeit angenommen, dass PH die dritthäufigste kardiovaskuläre Krankheitsgruppe nach koronarer Herzkrankheit und systemischem Bluthochdruck ist [Naeije, in: A. J. Peacock et al. (Eds.), Pulmonary Circulation. Diseases and their treatment, 3rd edition, Hodder Arnold Publ., 2011 , 3]. Die Einteilung der pulmonalen Hypertonie in verschiedene Gruppen gemäß der jeweiligen Ätiologie erfolgt seit 2008 nach der Dana Point-Klassifikation [D. Montana und G. Simonneau, in: A. J. Peacock et al. (Eds.), Pulmonary Cir- culation. Diseases and their treatment, 3rd edition, Hodder Arnold Publ., 2011 , 197-206].
Trotz aller Fortschritte in der Therapie der PH gibt es bisher keine Aussicht auf Heilung dieser schwerwiegenden Erkrankung. Auf dem Markt befindliche Therapien (z.B. Prostacyclin-Analoga, Endothelinrezeptor-Antagonisten, Phosphodiesterase-Inhibitoren) sind in der Lage, die Lebensqualität, die körperliche Belastbarkeit und die Prognose der Patienten zu verbessern. Hierbei handelt es sich um systemisch applizierte, primär hämodynamisch wirkende Therapieprinzipien, die den Gefäßtonus beeinflussen. Die Anwendbarkeit dieser Medikamente ist durch die z.T. gravierenden Nebenwirkungen und/oder aufwendigen Applikationsformen eingeschränkt. Der Zeitraum, über den unter einer spezifischen Monotherapie die klinische Situation der Patienten stabilisiert oder verbessert werden kann, ist begrenzt (z.B. aufgrund einer Toleranzentwicklung). Es erfolgt schließlich eine Therapieeskalation und somit eine Kombinationstherapie, bei der mehrere Medikamente gleichzeitig gegeben werden müssen. Zur Zeit sind diese Standardtherapeutika nur zur Behandlung der pulmonal-arteriellen Hypertonie (PAH) zugelassen. Bei sekundären For-
men der PH, wie z.B. PH-COPD, scheiterten diese Therapieprinzipien (z.B. Sildenafil, Bosentan) in klinischen Studien, da sie infolge einer unselektiven Vasodilatation zu einer Absenkung (Ent- sättigung) des arteriellen Sauerstoffgehalts bei den Patienten führten. Ursache hierfür ist wahrscheinlich eine ungünstige Beeinflussung der Ventilations-Perfusions-Anpassung innerhalb der Lunge bei heterogenen Lungenerkrankungen aufgrund der systemischen Gabe unselektiver Va- sodilatatoren [I. Blanco et at., Am. J. Respir. Crit. Care Med. 2010, 181, 270-278; D. Stolz et at., Eur. Respir. J. 2008, 32, 619-628].
Neue Kombinationstherapien sind eine der aussichtsreichsten zukünftigen Therapieoptionen zur Behandlung der pulmonalen Hypertonie. In diesem Zusammenhang ist die Erkundung neuer pharmakologischer Mechanismen zur Behandlung der PH von besonderem Interesse [Ghofrani et al., Herz 2005, 30, 296-302; E. B. Rosenzweig, Expert Opin. Emerging Drugs 2006, 11, 609-619; T. Ito et al., Curr. Med. Chem. 2007, 14, 719-733]. Vor allem solche neuen Therapieansätze, die mit den bereits auf dem Markt befindlichen Therapiekonzepten kombinierbar sind, könnten Grundlage einer effizienteren Behandlung sein und somit einen großen Vorteil für die Patienten bringen. Im Sinne der vorliegenden Erfindung schließt der Begriff pulmonale Hypertonie sowohl primäre als auch sekundäre Unterformen (NPAHPH) ein, wie sie nach der Dana Point-Klassifikation gemäß ihrer jeweiligen Ätiologie definiert worden sind [D. Montana und G. Simonneau, in: A. J. Peacock et al. (Eds.), Pulmonary Circulation. Diseases and their treatment, 3rd edition, Hodder Arnold Publ., 201 1 , 197-206; Hoeper et al., J. Am. Coli. Cardio!., 2009, 54 (1), Suppl. S, S85- S96]. Hierzu gehört insbesondere in Gruppe 1 die pulmonal-arterielle Hypertonie (PAH), zu der unter anderem die idiopathischen und die familiären Formen zählen (IPAH bzw. FPAH). Des weiteren umfasst PAH auch die persistierende pulmonale Hypertonie bei Neugeborenen sowie die assoziierte pulmonal-arterielle Hypertonie (APAH), welche assoziiert ist mit Kollagenosen, kongenitalen systemisch-pulmonalen Shuntvitien, portaler Hypertension, HIV-Infektionen, der Einnahme bestimmter Drogen und Medikamente (z.B. von Appetitzüglern), mit Erkrankungen mit einer signifikanten venösen/kapillären Beteiligung wie der pulmonal-venookklusiven Erkrankung und der pulmonal-kapillären Hämangiomatose, oder mit anderen Erkrankungen wie Schilddrüsenerkrankungen, Glykogenspeicherkrankheiten, Morbus Gaucher, hereditärer Teleangiektasie, Hämoglobinopathien, myeloproliferativen Erkrankungen und Splenektomie. In Gruppe 2 der Dana Point-Klassifikation werden PH-Patienten mit einer ursächlichen Linksherzerkrankung, wie ventrikulären, atrialen oder valvulären Erkrankungen, zusammengefasst. Gruppe 3 umfasst Formen der pulmonalen Hypertonie, die mit einer Lungenerkrankung, wie z.B. chronischobstruktiver Lungenerkrankung (COPD), interstitieller Lungenkrankheit (ILD), Lungenfibrose (IPF), und/oder einer Hypoxämie (z.B. Schlafapnoe-Syndrom, alveoläre Hypoventilation, chro- nische Höhenkrankheit, anlagebedingte Fehlbildungen) assoziiert sind. Zur Gruppe 4 zählen PH- Patienten mit chronisch-thrombotischen und/oder embolischen Erkrankungen, z.B. bei thrombo- embolischer Obstruktion von proximalen und distalen Lungenarterien (CTEPH) oder bei nicht-
thrombotischen Embolisierungen (z.B. infolge von Tumorerkrankungen, Parasiten, Fremdkörpern). Seltenere Formen der pulmonalen Hypertonie, wie z.B. bei Patienten mit Sarkoidose, His- tiozytose X oder Lymphangiomatose, sind in der Gruppe 5 zusammengefasst.
Beim Bronchiolitis obliterans-Syndrom (BOS) handelt es sich um eine chronische Abstoßungs- reaktion nach erfolgter Lungentransplantation. Innerhalb der ersten fünf Jahre nach Lungentransplantation sind ca. 50-60% aller Patienten, innerhalb der ersten neun Jahre über 90% der Patienten betroffen [Estenne et al., Am. J. Respir. Crit. Care Med. 2003, 766, 440-444]. Die Ursache der Erkrankung ist nicht geklärt. Trotz vieler Fortschritte bei der Behandlung von Transplantationspatienten haben sich die BOS-Fallzahlen in den vergangenen Jahren kaum verändert. Das BOS ist die wichtigste langfristige Komplikation bei Lungentransplantationen und gilt als Hauptgrund dafür, dass die Überlebensraten nach wie vor deutlich unter denen anderer Organtransplantationen liegen. Beim BOS handelt es sich um ein entzündliches Geschehen, das mit Veränderungen des Lungengewebes einhergeht, die vor allem die kleinen Atemwege betreffen. Die Schädigung und entzündlichen Veränderungen der Epithelzellen sowie der subepithelialen Strukturen der kleineren Atemwege führen aufgrund einer nicht effektiven Regeneration des Epithels und einer aberrierenden Gewebereparation zu einer exzessiven Fibroproliferation. Es kommt zur Vernarbung und schließlich Zerstörung der Bronchiolen sowie zu Pfropfen von Granulationsgewebe in den kleinen Atemwegen und Alveolen, gelegentlich auch mit vaskulärer Beteiligung. Die Diagnose wird aufgrund der Lungenfunktion gestellt. Beim BOS kommt es zu einer Verschlechterung des FEV1 im Vergleich zum Durchschnitt der zwei besten postoperativ gemessenen Werte. Gegenwärtig gibt es keine kurative Behandlung für BOS. Ein Teil der Patienten verbessert sich unter intensivierter Immunsuppression, bei den nicht darauf ansprechenden Patienten kommt es zu einer anhaltenden Verschlechterung, so dass eine erneute Transplantation (Retransplantation) indiziert ist.
Die chronisch-obstruktive Lungenerkrankung (COPD) ist eine langsam fortschreitende Lungen- erkrankung, die durch eine Behinderung der Atemströmung charakterisiert ist, welche durch ein Lungenemphysem und/oder eine chronische Bronchitis hervorgerufen wird. Die ersten Symptome der Erkrankung zeigen sich in der Regel ab dem vierten bis fünften Lebensjahrzehnt. In den darauffolgenden Lebensjahren verschlimmert sich häufig die Kurzatmigkeit und es manifestiert sich Husten, verbunden mit einem ausgiebigen und stellenweise eitrigen Auswurf und einer Stenose- Atmung bis hin zu einer Atemnot (Dyspnoe). COPD ist in erster Linie eine Krankheit von Rauchern: Rauchen ist verantwortlich für 90% aller COPD-Fälle und 80-90% aller COPD-Todesfälle. COPD ist ein großes medizinisches Problem und stellt weltweit die sechsthäufigste Todesursache dar. Von den über 45-jährigen Menschen sind ca. 4-6% betroffen. Obwohl die Behinderung der Atemströmung nur partiell und zeitlich befristet sein kann, ist COPD nicht heilbar. Behandlungsziel ist folglich eine Verbesserung der Lebensqualität, die Linderung der Symptome, die Verhinderung akuter Verschlechterungen und die Verlangsamung der fortschreitenden Beeinträchtigung der Lungenfunktion. Bestehende Pharmakotherapien, die sich seit den letzten zwei bis drei Jahrzehn-
ten kaum geändert haben, sind das Verwenden von Bronchodilatoren, um blockierte Atemwege zu öffnen, und in bestimmten Situationen Kortikosteroide, um die Entzündung der Lunge einzudämmen [P. J. Barnes, N. Engl. J. Med. 2000, 343, 269-280]. Die chronische Entzündung der Lunge, hervorgerufen durch Zigarettenrauch oder andere Reizstoffe, ist die treibende Kraft der Krank- heitsentwicklung. Der zugrunde liegende Mechanismus beinhaltet Immunzellen, die im Zuge der inflammatorischen Reaktion der Lunge Proteasen und verschiedene Zytokine ausschütten, die zu einem Lungenemphysem und Remodeling der Bronchien führen.
Die Aufgabe der vorliegenden Erfindung liegt in der Identifizierung und Bereitstellung neuer Substanzen, die potente, chemisch und metabolisch stabile, nicht-prostanoide Antagonisten des FP- Rezeptors darstellen und sich als solche zur Behandlung und/oder Prävention insbesondere von fibrotischen und inflammatorischen Erkrankungen eignen.
Unter anderem aus WO 95/32948-A1 , WO 96/02509-A1 , WO 97/19926-A1 und WO 2000/031038- A1 sind 2-Arylchinolin-4-carboxamide als IMK3- oder duale NK2/NK3-Antagonisten bekannt, welche sich zur Behandlung von Erkrankungen der Lunge und des Zentralnervensystems eignen. WO 2016/004035 offenbart 2-Arylchinolin-4-carboxamide als TSH-Rezeptor-Agonisten, welche zur Behandlung von Funktionsstörungen und malignen Veränderungen der Schilddrüse dienen können. In WO 2000/064877 werden Chinolin-4-carboxamid-Derivate beansprucht, welche als IMK3- Antagonisten zur Behandlung verschiedenartiger Erkrankungen, u.a. der Lunge und des Zentralnervensystems, eingesetzt werden können. In WO 2004/045614-A1 werden bestimmte Chinolin- carboxamide als Glucokinase-Liganden für die Behandlung von Diabetes beschrieben. In WO 2006/094237-A2 werden Chinolin-Derivate als Sirtuin-Modulatoren offenbart, die zur Behandlung verschiedenartiger Erkrankungen eingesetzt werden können. In WO 2011/153553-A2 werden verschiedene bicyclische Heteroaryl-Verbindungen als Kinase-Inhibitoren für die Behandlung insbesondere von Krebserkrankungen beansprucht. In EP 2 415 755-A1 sind unter anderem Chino- lin-Derivate beschrieben, die sich zur Behandlung von Erkrankungen eignen, welche mit der Aktivität des Plasminogen-Aktivator-lnhibitor-1 (PAI-1 ) assoziiert sind. In WO 2013/074059-A2 werden verschiedene Chinolin-4-carboxamid-Derivate aufgeführt, die als Inhibitoren von Cytosin- Deaminasen zur Verstärkung einer DNA-Transfektion von Zellen dienen können. In WO 2013/164326-A1 werden /V,3-Diphenylnaphthalin-1 -carboxamide als Agonisten des EP2-Prosta- glandin-Rezeptors zur Behandlung von Atemwegserkrankungen offenbart. In WO 2014/1 17090- A1 werden verschiedene 2-Arylchinolin-Derivate als Inhibitoren von Metalloenzymen beschrieben. In WO 2012/122370-A2 sind Chinolin-4-carboxamid-Derivate offenbart, die zur Behandlung von Autoimmun- und Krebserkrankungen eingesetzt werden können. In WO 2015/094912-A1 werden unter anderem substituierte /V,2-Diphenylchinolin-4-carboxamid-Derivate offenbart, die sich als An- tagonisten des Prostaglandin EP4-Rezeptors zur Behandlung von Arthritis und damit verbundenen Schmerzzuständen eignen. WO 2016/061280 offenbart Protein-Tyrosin-Phosphatase Modulatoren
mit einem 4-Amin-2-arylchinolin-Grundkörper, welche unter anderem zur Behandlung von Fettstoffwechselstörungen, Diabetes und Herz-Kreislauferkrankungen eingesetzt werden können.
Die vorliegende Erfindung betrifft Verbindungen der allgemeinen Formel (I)
Ar für Phenyl oder für Pyridyl steht,
wobei Phenyl bis zu vierfach und Pyridyl bis zu zweifach, jeweils gleich oder verschieden, mit Fluor, Chlor, mit bis zu dreifach mit Fluor substituiertem (Ci-C4)-Alkyl, mit bis zu vierfach mit Fluor substituiertem (C3-C4)-Cycloalkyl, mit bis zu dreifach mit Fluor substituiertem (Ci-C2)-Alkoxy, oder mit bis zu dreifach mit Fluor substituiertem (Ci-C2)-Alkylsulfanyl, substituiert sein können, oder wobei zwei Substituenten der Phenyl- oder Pyridylgruppe, falls sie an benachbarte Ringatome gebunden sind, gegebenenfalls so miteinander verbunden sind, dass sie zusammen eine Methylendioxy- oder Ethylendioxygruppe bilden, oder
wobei Phenyl bis zu fünffach mit Fluor substituiert sein kann,
Y für eine Bindung oder für eine Gruppe der Formel
#1-X-(CR10AR10B)k-#2
#1 für die Anknüpfstelle an das Kohlenstoffatom steht,
#2 für die Anknüpfstelle an die Carboxygruppe steht,
X für eine Bindung, -CH2-, -O-, -S(=0)m- oder -N(R11)- steht, worin
m 0, 1 oder 2 bedeutet, und
R11 Wasserstoff oder Methyl bedeutet,
R10A und R10B unabhängig voneinander für Wasserstoff, Fluor oder Methyl stehen, oder
R10A und R10B bilden zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, eine Cyclopropylgruppe,
k für 1 , 2, 3 oder 4 steht,
R1 für Halogen, bis zu fünffach mit Fluor substituiertes (Ci-C4)-Alkyl, bis zu dreifach mit Fluor substituiertes Methoxy, (Trifluormethyl)sulfanyl, Pentafluorsulfanyl, Trimethylsilyl, Ethinyl, Cyclopropyl, oder Cyclobutyl steht,
wobei Cyclopropyl und Cyclobutyl bis zu vierfach mit Fluor substituiert sein können,
R2, R3 und R4 unabhängig voneinander für Wasserstoff, Halogen oder bis zu dreifach mit Fluor substituiertes Methyl stehen,
R5 für Halogen, bis zu fünffach mit Fluor substituiertes (Ci-C4)-Alkyl, bis zu dreifach mit Fluor substituiertes Methoxy, Hydroxy, Methylsulfanyl, (Trifluormethyl)sulfanyl, Cyano, Ethenyl, Cyclopropyl oder Cyclobutyl steht,
wobei Cyclopropyl und Cyclobutyl bis zu vierfach mit Fluor substituiert sein können,
R6 für Phenyl, das bis zu dreifach, gleich oder verschieden, mit Fluor, Chlor, bis zu dreifach mit Fluor substituiertem Methyl und bis zu dreifach mit Fluor substituiertem Methoxy substituiert sein kann, oder für Thienyl, das ein- oder zweifach mit Methyl oder einfach mit Chlor oder Brom substituiert sein kann, oder für Thiazolyl oder Pyridyl steht,
R7A und R7B unabhängig voneinander für Wasserstoff oder Methyl stehen,
oder
R7A und R7B bilden zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, eine Cyclopropylgruppe,
R8 für Wasserstoff, Fluor, Methyl, Trifluormethyl, Ethyl oder Hydroxy steht,
R9 für Wasserstoff oder Methyl steht,
sowie ihre /V-Oxide, Salze, Solvate, Salze der /V-Oxide und Solvate der /V-Oxide und Salze.
Erfindungsgemäße Verbindungen sind die Verbindungen der Formel (I) und deren Salze, Solvate und Solvate der Salze, die von Formel (I) umfassten Verbindungen der nachfolgend ge- nannten Formeln und deren Salze, Solvate und Solvate der Salze sowie die von Formel (I) umfassten, nachfolgend als Ausführungsbeispiele genannten Verbindungen und deren Salze, Sol-
vate und Solvate der Salze, soweit es sich bei den von Formel (I) umfassten, nachfolgend genannten Verbindungen nicht bereits um Salze, Solvate und Solvate der Salze handelt.
Erfindungsgemäße Verbindungen sind ebenso /V-Oxide der Verbindungen der Formel (I) sowie deren Salze, Solvate und Solvate der Salze.
Als Salze sind im Rahmen der vorliegenden Erfindung physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen bevorzugt. Umfasst sind auch Salze, die für pharmazeutische Anwendungen selbst nicht geeignet sind, jedoch beispielsweise für die Isolierung, Reinigung oder Lagerung der erfindungsgemäßen Verbindungen verwendet werden können.
Physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen umfassen insbesonde- re die von üblichen Basen abgeleiteten Salze, wie beispielhaft und vorzugsweise Alkalimetallsalze (z.B. Natrium- und Kaliumsalze), Erdalkalisalze (z.B. Calcium- und Magnesiumsalze), Zinksalze sowie Ammoniumsalze abgeleitet von Ammoniak oder organischen Aminen mit 1 bis 16 C-Ato- men, wie beispielhaft und vorzugsweise Ethylamin, Diethylamin, Triethylamin, DIPEA, Monoetha- nolamin, Diethanolamin, Triethanolamin, Dimethylaminoethanol, Diethylaminoethanol, Tris(hydro- xymethyl)aminomethan, Cholin (2-Hydroxy-/V,/V,/V-trimethylethanaminium), Procain, Dicyclohexyl- amin, Dibenzylamin, /V-Methylmorpholin, /V-Methylpiperidin, Arginin, Lysin und 1 ,2-Ethylendiamin.
Darüber hinaus umfassen physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen auch Säureadditionssalze von Mineralsäuren, Carbonsäuren und Sulfonsäuren, z.B. Salze der Chlorwasserstoffsäure, Bromwasserstoffsäure, Schwefelsäure, Phosphorsäure, Me- thansulfonsäure, Ethansulfonsäure, Benzolsulfonsäure, Toluolsulfonsäure, Naphthalindisulfon- säure, Ameisensäure, Essigsäure, Trifluoressigsäure, Propionsäure, Bernsteinsäure, Fumarsäure, Maleinsäure, Milchsäure, Weinsäure, Äpfelsäure, Zitronensäure, Gluconsäure, Benzoesäure und Embonsäure.
Als Solvate werden im Rahmen der Erfindung solche Formen der erfindungsgemäßen Verbin- düngen bezeichnet, welche in festem oder flüssigem Zustand durch Koordination mit Lösungsmittelmolekülen einen Komplex bilden. Hydrate sind eine spezielle Form der Solvate, bei denen die Koordination mit Wasser erfolgt. Als Solvate sind im Rahmen der vorliegenden Erfindung Hydrate bevorzugt.
Die erfindungsgemäßen Verbindungen können in Abhängigkeit von ihrer Struktur in unterschied- liehen stereoisomeren Formen existieren, d.h. in Gestalt von Konfigurationsisomeren oder gegebenenfalls auch als Konformationsisomere (Enantiomere und/oder Diastereomere, einschließlich solcher bei Atropisomeren). Die vorliegende Erfindung umfasst deshalb die Enantiomere und Diastereomere sowie ihre jeweiligen Mischungen. Aus solchen Mischungen von Enantiomeren und/ oder Diastereomeren lassen sich die stereoisomer einheitlichen Bestandteile in bekannter Weise isolieren. Vorzugsweise werden hierfür chromatographische Verfahren angewandt, insbesondere die HPLC-Chromatographie an achiralen bzw. chiralen Trennphasen. Im Falle von
Carbonsäuren als Zwischen- oder Endprodukten kann alternativ auch eine Trennung über dia- stereomere Salze mit Hilfe chiraler Amin-Basen erfolgen.
Der Begriff "enantiomerenrein" wird im Rahmen der vorliegenden Erfindung dahingehend verstanden, dass die betreffende Verbindung hinsichtlich der Absolutkonfiguration der chiralen Zentren in einem Enantiomerenüberschuss von mehr als 95%, bevorzugt von mehr als 98% vorliegt. Der Enantiomerenüberschuss (engl, enantiomeric excess, ee-Wert) wird hierbei durch Auswertung des Chromatogramms einer HPLC-Analyse an chiraler Phase nach der folgenden Formel berechnet:
Enantiomer 1 (Flächenprozent)— Enantiomer 2 (Flächenprozent)
ee = — x 100%
Enantiomer 1 (Flächenprozent) + Enantiomer 2 (Flächenprozent)
Sofern die erfindungsgemäßen Verbindungen in tautomeren Formen vorkommen können, um- fasst die vorliegende Erfindung sämtliche tautomere Formen.
Die vorliegende Erfindung umfasst auch alle geeigneten isotopischen Varianten der erfindungsgemäßen Verbindungen. Unter einer isotopischen Variante einer erfindungsgemäßen Verbindung wird hierbei eine Verbindung verstanden, in welcher mindestens ein Atom innerhalb der erfindungsgemäßen Verbindung gegen ein anderes Atom der gleichen Ordnungszahl, jedoch mit einer anderen Atommasse als der gewöhnlich oder überwiegend in der Natur vorkommenden Atommasse ausgetauscht ist. Beispiele für Isotope, die in eine erfindungsgemäße Verbindung inkorporiert werden können, sind solche von Wasserstoff, Kohlenstoff, Stickstoff, Sauerstoff, Phosphor, Schwefel, Fluor, Chlor, Brom und lod, wie 2H (Deuterium), 3H (Tritium), 13C, 14C, 15N, 170, 180, 32P, 33P, 33S, ^S, 35S, 36S, 18F, 36CI, 82Br, 123l, 124l, 129l und 131l. Bestimmte isotopische Varianten einer er- findungsgemäßen Verbindung, wie insbesondere solche, bei denen ein oder mehrere radioaktive Isotope inkorporiert sind, können von Nutzen sein beispielsweise für die Untersuchung des Wirkmechanismus oder der Wirkstoff-Verteilung im Körper; aufgrund der vergleichsweise leichten Herstell- und Detektierbarkeit sind hierfür insbesondere mit 3H- oder 14C-lsotopen markierte Verbindungen geeignet. Darüber hinaus kann der Einbau von Isotopen, wie beispielsweise von Deu- terium, zu bestimmten therapeutischen Vorteilen als Folge einer größeren metabolischen Stabilität der Verbindung führen, wie beispielsweise zu einer Verlängerung der Halbwertszeit im Körper oder zu einer Reduktion der erforderlichen Wirkdosis; solche Modifikationen der erfindungsgemäßen Verbindungen können daher gegebenenfalls auch eine bevorzugte Ausführungsform der vorliegenden Erfindung darstellen. Isotopische Varianten der erfindungsgemäßen Verbindungen können nach allgemein gebräuchlichen, dem Fachmann bekannten Verfahren hergestellt werden, so beispielsweise nach den weiter unten beschriebenen Methoden und den bei den Ausführungsbeispielen wiedergegebenen Vorschriften, indem hierbei entsprechende isotopische Modifikationen der jeweiligen Reagentien und/oder Ausgangsverbindungen eingesetzt werden.
Außerdem umfasst die vorliegende Erfindung auch Prodrugs der erfindungsgemäßen Verbindungen. Der Begriff "Prodrugs" bezeichnet hierbei Verbindungen, welche selbst biologisch aktiv oder inaktiv sein können, jedoch während ihrer Verweilzeit im Körper auf beispielsweise metabolischem oder hydrolytischem Wege zu erfindungsgemäßen Verbindungen umgesetzt werden. Insbesondere umfasst die vorliegende Erfindung als Prodrugs hydrolysierbare Ester-Derivate der erfindungsgemäßen Carbonsäuren der Formel (I). Hierunter werden Ester verstanden, die in physiologischen Medien, unter den Bedingungen der im weiteren beschriebenen biologischen Tests und insbesondere in vivo auf enzymatischem oder chemischem Wege zu den freien Carbonsäuren, als den biologisch hauptsächlich aktiven Verbindungen, hydrolysiert werden können. Als solche Ester werden (Ci-C4)-Alkylester der Formel (IV), in welchen die Alkylgruppe geradkettig o- der verzweigt sein kann, bevorzugt. Besonders bevorzugt sind Methyl-, Ethyl- oder ie/f-Butylester.
Im Rahmen der vorliegenden Erfindung haben die Substituenten, soweit nicht anders spezifiziert, die folgende Bedeutung:
Der Begriff "Halogen oder„Halogenatom" bezeichnet ein Fluor-, Chlor-, Brom- oder lodatom. Der Begriff „Ci-C4-Alkyl" bezeichnet eine geradkettige oder verzweigte gesättigte einwertige Kohlenwasserstoffgruppe mit 1 , 2, 3 oder 4 Kohlenstoffatomen, z. B. eine Methyl-, Ethyl-, Propyl- , Isopropyl-, Butyl-, se/ -Butyl-, Isobutyl- oder ie/f-Butylgruppe oder ein Isomer davon.
Der Begriff „C3-C4-Cycloalkyl" bezieht sich auf einen gesättigten einwertigen mono- oder bicycli- schen Kohlenwasserstoffring mit 3 oder 4 Kohlenstoffatomen. Bei der C3-C4-Cycloalkylgruppe handelt es sich zum Beispiel um einen monocyclischen Kohlenwasserstoffring, z. B. eine Cyc- lopropyl- oder Cyclobutylgruppe.
Der Begriff„Ci-C2-Alkoxy" bezeichnet eine geradkettige oder verzweigte gesättigte einwertige Gruppe der Formel (Ci-C2-Alkyl)-0-, in welcher der Begriff„Ci-C2-Alkyl" wie oben definiert ist, z. B. eine Methoxy- oder Ethoxygruppe oder ein Isomer davon.
Der Begriff„Ci-C2-Alkylsulfanyl" bezeichnet eine geradkettige oder verzweigte gesättigte einwertige Gruppe der Formel (Ci-C2-Alkyl)-S-, in welcher der Begriff„Ci-C2-Alkyl" wie oben definiert ist, z. B. eine Methylsulfanyl- oder Ethylsulfanylgruppe.
Ein Oxosubstituent steht im Rahmen der Erfindung für ein Sauerstoffatom, das über eine Doppelbindung an ein Kohlenstoffatom gebunden ist.
Der Begriff„C1-C4" bezieht sich, so wie er in diesem Text verwendet wird, z. B. im Zusammenhang mit der Definition von „Ci-C4-Alkyl" auf eine Alkylgruppe mit einer begrenzten Anzahl an Kohlenstoffatomen von 1 bis 3, d. h. 1 , 2, 3 oder 4 Kohlenstoffatomen.
Wird ein Bereich von Werten aufgeführt, so soll dieser jeden einzelnen Wert und Unterbereiche innerhalb des Bereichs umfassen.
„C1-C4" beispielsweise soll C1 , C2, C3, C4, C1-C4, C1-C3, C1-C2, C2-C4, C2-C3 und C3-C4 umfassen.
Im Rahmen der vorliegenden Erfindung gilt, dass für alle Reste, die mehrfach auftreten, deren Bedeutung unabhängig voneinander ist. Wenn Reste in den erfindungsgemäßen Verbindungen substituiert sind, können die Reste, soweit nicht anders spezifiziert, ein- oder mehrfach substitu- iert sein. Eine Substitution mit einem oder mit zwei gleichen oder verschiedenen Substituenten ist bevorzugt. Besonders bevorzugt ist die Substitution mit einem Substituenten.
Bevorzugt im Rahmen der vorliegenden Erfindung sind Verbindungen der Formel (I), in welcher
Ar für Phenyl oder für 2-Pyridyl steht,
wobei Phenyl bis zu vierfach mit Fluor oder bis zu dreifach, gleich oder verschieden, mit Fluor, Chlor, Methyl, Trifluormethyl, Difluormethyl, Methoxy, Difluormethoxy, Trifluorme- thoxy, oder Ethoxy, substituiert sein kann, oder wobei zwei Substituenten der Phenylgrup- pe, falls sie an benachbarte Ringatome gebunden sind, gegebenenfalls so miteinander verbunden sind, dass sie zusammen eine Methylendioxygruppe bilden, und
wobei 2-Pyridyl bis zu zweifach, gleich oder verschieden, mit Chlor oder Methoxy substitu- iert sein kann,
Y für eine Bindung oder für eine Gruppe der Formel
#1-X-(CR10AR10B)k-#2
steht, wobei
#1 für die Anknüpfstelle an das Kohlenstoffatom steht,
#2 für die Anknüpfstelle an die Carboxygruppe steht,
X für -CH2-, -O-, -S(=0)m- oder -N(R11)- steht, worin
m 0 oder 2 bedeutet, und
R11 Wasserstoff oder Methyl bedeutet,
R10A und R10B unabhängig voneinander für Wasserstoff, Fluor oder Methyl stehen, k für 1 , 2 oder 3 steht,
R1 für Chlor, Brom, lod, Methyl, Isopropyl, ie/f-Butyl, Difluormethyl, Trifluormethyl, Trifluorme- thoxy, (Trifluormethyl)sulfanyl, Trimethylsilyl, Ethinyl, Cyclopropyl oder Cyclobutyl steht,
R2 für Wasserstoff steht,
R3 und R4 unabhängig voneinander für Wasserstoff, Chlor oder Methyl stehen,
R5 für Fluor, Chlor, Brom, lod, Methyl, Ethyl, Propyl, Monofluormethyl, Difluormethyl, Trifluormethyl, Methoxy, Trifluormethoxy, Hydroxy, Methylsulfanyl oder Cyclopropyl steht,
und
R6 für Phenyl, das ein- oder zweifach, gleich oder verschieden, mit Fluor oder Chlor oder einfach mit Methyl, Trifluormethyl, Methoxy oder Trifluormethoxy substituiert sein kann, oder für Thienyl, das ein- oder zweifach mit Methyl oder einfach mit Chlor oder Brom substituiert sein kann, steht,
R7A für Wasserstoff oder Methyl steht,
R7B für Wasserstoff steht,
R8 für Wasserstoff, Fluor, Methyl, Ethyl oder Hydroxy steht,
R9 für Wasserstoff steht,
sowie ihre Salze, Solvate und Solvate der Salze.
Bevorzugt im Rahmen der vorliegenden Erfindung sind Verbindungen der Formel (I), in welcher Ar für Phenyl steht,
wobei Phenyl bis zu vierfach mit Fluor oder bis zu dreifach, gleich oder verschieden, mit Fluor, Chlor, Methyl, Trifluormethyl, Difluormethyl, Methoxy, Difluormethoxy oder Trifluor- methoxy substituiert sein kann,
Y für eine Gruppe der Formel
#1-(CH2)n-#2
steht, wobei
#1 für die Anknüpfstelle an das Kohlenstoffatom steht,
#2 für die Anknüpfstelle an die Carboxygruppe steht,
n für 1 , 2 oder 3 steht,
R1 für Brom oder Ethinyl steht,
R2, R3 und R4 jeweils für Wasserstoff stehen,
R5 für Chlor oder Methyl steht,
und
R6 für Phenyl, das einfach mit Fluor substituiert sein kann, steht,
R7A und R7B jeweils für Wasserstoff stehen,
R8 für Wasserstoff oder Methyl steht,
R9 für Wasserstoff steht,
sowie ihre Salze, Solvate und Solvate der Salze.
Besonders bevorzugt im Rahmen der vorliegenden Erfindung sind Verbindungen der Formel (I), in welcher
Ar für Phenyl steht,
wobei Phenyl bis zu vierfach mit Fluor oder bis zu dreifach, gleich oder verschieden, mit Flu- or, Chlor, Methyl, Trifluormethyl, Difluormethoxy oder Trifluormethoxy, substituiert sein kann,
Y für eine Gruppe der Formel
#1-CH2CH2-#2
steht, wobei
#1 für die Anknüpfstelle an das Kohlenstoffatom steht,
#2 für die Anknüpfstelle an die Carboxygruppe steht,
R1 für Brom oder Ethinyl steht,
R2, R3, R4 jeweils für Wasserstoff stehen,
R5 für Methyl oder Chlor steht,
R6 für Phenyl steht,
R7A, R7B, R8 und R9 jeweils für Wasserstoff stehen,
sowie ihre Salze, Solvate und Solvate der Salze.
Eine besondere Ausführungsform der vorliegenden Erfindung umfasst Verbindungen der Formel (I), in welcher
R1 für Brom steht, und
R2, R3 und R4 jeweils für Wasserstoff stehen,
sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
Eine weitere besondere Ausführungsform der vorliegenden Erfindung umfasst Verbindungen der Formel (I), in welcher
R1 für Ethinyl steht, und
R2, R3 und R4 jeweils für Wasserstoff stehen,
sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
Eine weitere besondere Ausführungsform der vorliegenden Erfindung umfasst Verbindungen der Formel (I), in welcher
R5 für Methyl oder Chlor steht,
sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
Eine weitere besondere Ausführungsform der vorliegenden Erfindung umfasst Verbindungen der Formel (I), in welcher
R5 für Methyl steht,
sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze. Eine weitere besondere Ausführungsform der vorliegenden Erfindung umfasst Verbindungen der Formel (I), in welcher
R6 für Phenyl, das einfach mit Fluor substituiert sein kann, steht,
Eine ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
Eine weitere besondere Ausführungsform der vorliegenden Erfindung umfasst Verbindungen der Formel (I), in welcher
R6 für Phenyl steht,
sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
Eine weitere besondere Ausführungsform der vorliegenden Erfindung umfasst Verbindungen der Formel (I), in welcher
Y für eine Bindung steht,
sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
Eine weitere besondere Ausführungsform der vorliegenden Erfindung umfasst Verbindungen der Formel (I), in welcher
Y für eine Gruppe der Formel
#1-CH2-#2
steht, wobei
#1 für die Anknüpfstelle an das Kohlenstoffatom steht,
#2 für die Anknüpfstelle an die Carboxygruppe steht,
sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze. Eine weitere besondere Ausführungsform der vorliegenden Erfindung umfasst Verbindungen der Formel (I), in welcher
Y für eine Gruppe der Formel
#1-CH2CH2-#2
steht, wobei
#1 für die Anknüpfstelle an das Kohlenstoffatom steht,
#2 für die Anknüpfstelle an die Carboxygruppe steht,
sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
Eine weitere besondere Ausführungsform der vorliegenden Erfindung umfasst Verbindungen der Formel (I), in welcher
X für -O- steht,
sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
Eine weitere besondere Ausführungsform der vorliegenden Erfindung umfasst Verbindungen der Formel (I), in welcher
R8 für Wasserstoff oder Methyl steht,
sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
Eine weitere besondere Ausführungsform der vorliegenden Erfindung umfasst Verbindungen der Formel (I), in welcher
R7A für Methyl steht,
R7B für Wasserstoff steht,
sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
Eine weitere besondere Ausführungsform der vorliegenden Erfindung umfasst Verbindungen der Formel (I), in welcher
Ar für Phenyl steht,
wobei Phenyl bis zu vierfach mit Fluor oder bis zu dreifach, gleich oder verschieden, mit Fluor, Chlor, Methyl, Trifluormethyl, Difluormethyl, Methoxy, Difluormethoxy, Trifluorme- thoxy, oder Ethoxy, substituiert sein kann, oder wobei zwei Substituenten der Phenylgrup- pe, falls sie an benachbarte Ringatome gebunden sind, gegebenenfalls so miteinander verbunden sind, dass sie zusammen eine Methylendioxygruppe bilden,
sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze. Eine weitere besondere Ausführungsform der vorliegenden Erfindung umfasst Verbindungen der Formel (I), in welcher
Ar für Phenyl steht,
wobei Phenyl bis zu vierfach mit Fluor oder bis zu dreifach, gleich oder verschieden, mit Fluor, Chlor, Methyl, Trifluormethyl, Difluormethoxy oder Trifluormethoxy, substituiert sein kann, sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
Eine weitere besondere Ausführungsform der vorliegenden Erfindung umfasst Verbindungen der Formel (I), in welcher
Ar für 2-Pyridyl steht,
wobei 2-Pyridyl bis zu zweifach, gleich oder verschieden, mit Chlor oder Methoxy substitu- iert sein kann,
sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
Eine weitere besondere Ausführungsform der vorliegenden Erfindung umfasst Verbindungen der Formel (I), in welcher
R2, R3 und R4 unabhängig voneinander für Wasserstoff stehen,
sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
Eine weitere besondere Ausführungsform der vorliegenden Erfindung umfasst Verbindungen der Formel (I), in welcher
R1 für Brom steht,
R2, R3, R4 jeweils für Wasserstoff stehen,
R5 für Methyl steht,
R6 für Phenyl steht,
sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
Eine weitere besondere Ausführungsform der vorliegenden Erfindung umfasst Verbindungen der Formel (I), in welcher
R1 für Ethinyl steht,
R2, R3, R4 jeweils für Wasserstoff stehen,
R5 für Methyl steht,
R6 für Phenyl steht,
sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze. Die in den jeweiligen Kombinationen bzw. bevorzugten Kombinationen von Resten im einzelnen angegebenen Reste-Definitionen werden unabhängig von den jeweiligen angegebenen Kombinationen der Reste beliebig auch durch Reste-Definitionen anderer Kombinationen ersetzt.
Ganz besonders bevorzugt sind Kombinationen von zwei oder mehreren der oben genannten Vorzugsbereiche.
Die als bevorzugt, besonders bevorzugt und ganz besonders bevorzugt genannten Restedefinitionen gelten sowohl für die Verbindungen der Formel (I) als auch in entsprechender Weise für
alle Zwischenprodukte.
Weiterer Gegenstand der Erfindung ist ein Verfahren zur Herstellung der erfindungsgemäßen Verbindungen der Formel (I), dadurch gekennzeichnet, dass man eine Verbindung der Formel (II)
in welcher R1, R2, R3, R4, R5 und R6 die oben angegebenen Bedeutungen haben,
in einem ersten Schritt
[A] unter Aktivierung der Carbonsäure-Funktion mit einer Amin-Verbindung der Formel (lll-A) oder (III-B)
in welcher R7A, R7B, R8, R9 und Ar die oben angegebenen Bedeutungen haben,
und
T für eine Ester-Schutzgruppe, insbesondere (Ci-C4)-Alkyl, steht,
umsetzt,
und gegebenenfalls in einem nachfolgenden Schritt
[B] den Ester-Rest T einer aus Schritt [A] nach Umsetzung mit einer Amin-Verbindung (III-B) erhaltenen Verbindung der Formel (IV)
(iv),
in welcher R
1, R
2, R
3, R
4, R
5, R
6, R
7A, R
7B, R
8, R
9 und Ar die oben angegebenen Bedeutungen haben,
und
T für eine Ester-Schutzgruppe, insbesondere (Ci-C4)-Alkyl, steht,
abspaltet,
und die so erhaltenen Verbindungen der Formel (I)
in welcher R1, R2, R3, R4, R5, R6, R7A, R7B, R8, R9 und Ar die oben angegebenen Bedeutungen haben,
gegebenenfalls in ihre Enantiomere und/oder Diastereomere trennt und/oder mit den entsprechenden (i) Lösungsmitteln und/oder (ii) Säuren oder Basen in ihre Solvate, Salze und/oder Sol- vate der Salze überführt.
Die Kupplungsreaktion (II) + (lll-A) -> (I) und (II) + (III-B) -> (IV) [Amid-Bildung] kann entweder auf direktem Weg mit Hilfe eines Kondensations- oder Aktivierungsmittels oder über die Zwischenstufe eines aus (II) erhältlichen Carbonsäurechlorids oder Carbonsäureimidazolids erfolgen.
Als solche Kondensations- oder Aktivierungsmittel eignen sich beispielsweise Carbodiimide wie Λ/,Λ/'-Diethyl-, Λ/,Λ/'-Dipropyl-, Λ/,Λ/'-Diisopropyl-, Λ/,Λ/'-Dicyclohexylcarbodiimid (DCC) oder /V-(3-Di- methylaminopropyl)-/V'-ethylcarbodiimid-Hydrochlorid (EDC), Phosgen-Derivate wie Λ/,Λ/'-Carbo- nyldiimidazol (CDI) oder Isobutylchlorformiat, 1 ,2-Oxazolium-Verbindungen wie 2-Ethyl-5-phenyl- 1 ,2-oxazolium-3-sulfat oder 2-ie/f-Butyl-5-methylisoxazolium-perchlorat, Acylamino-Verbindungen wie 2-Ethoxy-1-ethoxycarbonyl-1 ,2-dihydrochinolin, α-Chlorenamine wie 1-Chlor-/V,/V,2-trimethyl- prop-1-en-1-amin, 1 ,3,5-Triazin-Derivate wie 4-(4,6-Dimethoxy-1 ,3,5-triazin-2-yl)-4-methylmorpholi- niumchlorid, Phosphor-Verbindungen wie n-Propanphosphonsäureanhydrid (PPA), Cyanophos- phonsäurediethylester, Diphenylphosphorylazid (DPPA), Bis-(2-oxo-3-oxazolidinyl)-phosphoryl- Chlorid, Benzotriazol-1-yloxy-tris(dimethylamino)phosphonium-hexafluorophosphat oder Benzotri- azol-1-yloxy-tris(pyrrolidino)phosphonium-hexafluorophosphat (PyBOP), oder Uronium-Verbin- dungen wie 0-(Benzotriazol-1-yl)-/V,/V,/V',/V'-tetramethyluronium-tetrafluoroborat (TBTU), 0-(Ben-
zotriazol-1-yl)-/V,/V,/V /V^etramethyluronium-hexafluorophosphat (HBTU), 0-(1 /-/-6-Chlorbenzotri- azol-1-yl)-1 ,1 ,3,3-tetramethyluronium-tetrafluoroborat (TCTU), 0-(7-Azabenzotriazol-1-yl)-/V,/V,- Λ/',Λ/'-tetramethyluronium-hexafluorophosphat (HATU) oder 2-(2-Oxo-1-(2H)-pyridyl)-1 ,1 ,3,3-tetra- methyluronium-tetrafluoroborat (TPTU), gegebenenfalls in Kombination mit weiteren Hilfsstoffen wie 1-Hydroxybenzotriazol (HOBt) oder /V-Hydroxysuccinimid (HOSu), sowie als Basen Alkalicar- bonate, z.B. Natrium- oder Kaliumcarbonat, oder tertiäre Aminbasen wie Triethylamin, /V-Methyl- morpholin (NMM), /V-Methylpiperidin (NMP), DIPEA, Pyridin oder 4-/V,/V-Dimethylaminopyridin (DMAP). Als Kondensations- oder Aktivierungsmittel bevorzugt eingesetzt wird 0-(7-Azabenzotria- zol-1-yl)-/V,/V,/V',/V'-tetramethyluronium-hexafluorophosphat (HATU) in Kombination mit DIPEA. Bei zweistufiger Reaktionsführung über die aus (II) erhältlichen Carbonsäurechloride oder Car- bonsäureimidazolide wird die Kupplung mit der Amin-Komponente (lll-A) / (III-B) in Gegenwart einer üblichen Base durchgeführt, wie beispielsweise Natrium- oder Kaliumcarbonat, Triethylamin, DIPEA, /V-Methylmorpholin (NMM), /V-Methylpiperidin (NMP), Pyridin, 2,6-Dimethylpyridin, 4-/V,/V-Dimethylaminopyridin (DMAP), 1 ,8-Diazabicyclo[5.4.0]undec-7-en (DBU), 1 ,5-Diaza- bicyclo[4.3.0]non-5-en (DBN), Natrium- oder Kaliummethanolat, Natrium- oder Kaliumethanolat, Natrium- oder Kalium-ie/f-butylat oder Natrium- oder Kaliumhydrid. Bevorzugt wird Triethylamin oder DIPEA als Base eingesetzt.
Bevorzugte Kupplungsmethode ist die direkte Umsetzung von (II) mit der Amin-Verbindung (III) mit Hilfe eines Kondensations- oder Aktivierungsmittels.
Inerte Lösungsmittel für die genannten Kupplungsreaktionen sind - je nach eingesetztem Verfahren - beispielsweise Ether wie Diethylether, Diisopropylether, Methyl-ierf-butylether, Tetrahydro- furan, 1 ,4-Dioxan, 1 ,2-Dimethoxyethan oder Bis(2-methoxyethyl)ether, Kohlenwasserstoffe wie Benzol, Toluol, Xylol, Pentan, Hexan oder Cyclohexan, Halogenkohlenwasserstoffe wie Dichlor- methan, Trichlormethan, Tetrachlormethan, 1 ,2-Dichlorethan, Trichlorethylen oder Chlorbenzol, oder polar-aprotische Lösungsmittel wie Aceton, Methylethylketon, Ethylacetat, Acetonitril, Butyro- nitril, Pyridin, Dimethylsulfoxid (DMSO), /V,/V-Dimethylformamid (DMF), Λ/,Λ/'-Dimethylpropylen- harnstoff (DMPU) oder /V-Methylpyrrolidinon (NMP). Auch können Gemische solcher Lösungsmittel eingesetzt werden. Bevorzugt wird /V,/V-Dimethylformamid verwendet. Die Kupplungen werden im Allgemeinen in einem Temperaturbereich von 0°C bis +130°C, bevorzugt bei +20°C bis +80°C durchgeführt.
Die Carbonsäureimidazolide selbst sind nach bekanntem Verfahren durch Umsetzung von (II) mit Λ/,Λ/'-Carbonyldiimidazol (CDI) bei erhöhter Temperatur (+60°C bis +150°C) in einem entsprechend höhersiedenden Lösungsmittel wie /V,/V-Dimethylformamid (DMF) erhältlich. Die Herstellung der Carbonsäurechloride geschieht auf übliche Weise durch Behandlung von (II) mit Thionylchlorid oder Oxalylchlorid in einem inerten Lösungsmittel wie Dichlormethan.
Als Ester-Schutzgruppe T eignen sich im Allgemeinen alle dem Fachmann bekannten Schutzgruppen, beispielsweise geeignet substituiertes Methyl, wie Methylthiomethyl (MTM), Tetrahyd- ropyranyl (THP), 2-(Trimethylsilyl)ethoxymethyl (SEM), Benzyloxymethyl (BOM), Phenacyl und /V-Phthalimidomethyl, geeignet 2-substituiertes Ethyl, wie 4-Methylphenylsulfonylethyl (TSE), 2,2,2-Trichlorethyl, 2-(Trimethylsilyl)ethyl und 2-(2'-Pyridyl)ethyl (PET), Allyl, Benzyl, geeignet substituiertes Benzyl, wie Diphenylmethyl (DPM), Bis(o/f/?o-nitrophenyl)methyl, 9-Anthrylmethyl, 2,4,6-Trimethylbenzyl, 4-Brombenzyl, 4-Methoxybenzyl (PMB), Piperonyl und geeignet substituiertes Silyl, wie Triethylsilyl (TES), ie/f-Butyldimethylsilyl (TBDMS) und Di-ie/f-butylmethylsilyl (DTBMS), insbesondere und bevorzugt wird im erfindungsgemäßen Verfahren (Ci-C4)-Alkyl als Ester-Schutzgruppe T verwendet.
Die Abspaltung der Ester-Schutzgruppe T im Verfahrensschritt (IV) -^- (I) wird nach üblichen Methoden durchgeführt, indem man den Ester in einem inerten Lösungsmittel mit einer Säure oder Base behandelt, wobei bei letzterer Variante das zunächst entstehende Salz der Carbonsäure durch nachfolgende Behandlung mit Säure in die freie Carbonsäure überführt wird. Im Falle der ie/f-Butylester erfolgt die Esterspaltung vorzugsweise mit einer Säure. Methyl- und Ethylester werden bevorzugt mittels einer Base gespalten. Benzylester können alternativ auch durch Hydrierung (Hydrogenolyse) in Gegenwart eines geeigneten Katalysators, wie beispielsweise Palladium auf Aktivkohle, abgespalten werden. Silylester können durch Behandlung mit Säuren oder Fluoriden, z.B. Tetrabutylammoniumfluorid gespalten werden.
Als inerte Lösungsmittel eignen sich für diese Reaktionen Wasser und die für eine Esterspaltung üblichen organischen Lösungsmittel. Hierzu zählen insbesondere Alkohole wie Methanol, Ethanol, n-Propanol, Isopropanol, n-Butanol oder ie/f-Butanol, Ether wie Diethylether, Tetrahydrofuran, 1 ,4- Dioxan oder 1 ,2-Dimethoxyethan, oder andere Lösungsmittel wie Dichlormethan, Acetonitril, N,N- Dimethylformamid oder Dimethylsulfoxid. Ebenso ist es möglich, Gemische dieser Lösungsmittel einzusetzen. Im Falle einer basischen Ester-Hydrolyse werden bevorzugt Gemische von Wasser mit Tetrahydrofuran, 1 ,4-Dioxan, Methanol und/oder Ethanol verwendet. Im Falle der Umsetzung mit Trifluoressigsäure wird bevorzugt Dichlormethan und im Falle der Umsetzung mit Chlorwasserstoff bevorzugt 1 ,4-Dioxan, jeweils unter wasserfreien Bedingungen, verwendet.
Als Basen sind die für eine Hydrolyse-Reaktion üblichen anorganischen Basen geeignet. Hierzu gehören insbesondere Alkali- oder Erdalkalihydroxide wie beispielsweise Lithium-, Natrium-, Kalium- oder Bariumhydroxid, oder Alkali- oder Erdalkalicarbonate wie Natrium-, Kalium- oder Calciumcarbonat. Bevorzugt werden wässrige Lithiumhydroxid-Lösung oder Natriumhydroxid- Lösung (Natronlauge) eingesetzt.
Als Säuren eignen sich für die Esterspaltung im Allgemeinen Schwefelsäure, Chlorwas- serstoff/Salzsäure, Bromwasserstoff/Bromwasserstoffsäure, Phosphorsäure, Essigsäure, Trifluoressigsäure, Toluolsulfonsäure, Methansulfonsäure oder Trifluormethansulfonsäure oder deren
Gemische gegebenenfalls unter Zusatz von Wasser. Bevorzugt werden Chlorwasserstoff oder Trifluoressigsäure verwendet.
Die Esterspaltung wird in der Regel in einem Temperaturbereich von -20°C bis +100°C, bevorzugt bei 0°C bis +80°C durchgeführt.
Verbindungen der Formel (I) lassen sich nach üblichen Methoden in ihre korrespondierenden, von Basen abgeleiteten Salze überführen.
Von üblichen Basen abgeleitete Salze von Verbindungen der Formel (I) lassen sich zum Beispiel durch Zugabe von Basen, wie Alkali- und Erdalkalihydroxid-Lösungen zu Verbindungen der Formel (I) darstellen. Bevorzugt werden wässrige Hydroxidlösungen (z.B. Natriumhydroxid- Lösung) verwendet. Die wässrigen Hydroxidlösungen (z.B. Kaliumhydroxid-Lösung) können auch in situ generiert werden, indem eine in einem organischen Lösungsmittel gelöste metallorganische Verbindung (z.B. Kalium-ie/f-butylat-Lösung in THF) mit Wasser oder einer wässrigen Lösung vermischt wird. Die betreffenden, von Basen abgeleiteten Salze lassen sich auch durch Zugabe von Basen, wie Alkali- und Erdalkalihydroxid-Lösungen, zu Esterverbindungen der For- mel (IV) darstellen, indem zunächst die Abspaltung der Estergruppe T erfolgt, und das betreffende Salz in situ, d.h. ohne Isolierung der freien Carbonsäure direkt gebildet wird. Bevorzugte Estergruppen T hinsichtlich der Abspaltung durch Basen sind die Methyl- und die Ethylgruppe.
Die Verbindungen der Formel (II) können in Abhängigkeit vom jeweiligen Substitutionsmuster dadurch hergestellt werden, dass man in Analogie zu literaturbekannten Verfahren (s.a. WO 2016/146602 A1 , S. 26 bis 32) entweder ein Isatin-Derivat der Formel (V) in einer Säure- oder Base-vermittelten Kondensationsreaktion mit einer Ketomethylen-Verbindung der Formel (VI) zur Verbindung der Formel (VII) umsetzt (Schema 1 ) oder o/f/?o-Aminophenylessigsäureester der Formel (VIII) in einer Säure-induzierten Kondensationsreaktion mit einer Diketo-Verbindung der Formel (IX) zu einer Verbindung der Formel (Vll-A) umsetzt (Schema 2).
Schema 1 :
Die Kondensation des Isatin-Derivats der Formel (V) mit der Ketomethylen-Verbindung der Formel (VI) zur Chinolin-4-carbonsäure der Formel (VII) (Schema 1 ) kann durch Erhitzen der Reak- tanden in Gegenwart einer wässrigen Säure, wie Schwefelsäure oder konzentrierte Salzsäure, oder in Gegenwart einer wässrigen Base, wie Natron- oder Kalilauge, erzielt werden. Bei Verwendung einer Säure wird für die Umsetzung vorzugsweise Essigsäure als Lösungsmittel eingesetzt; bei basischer Reaktionsführung wird bevorzugt ein alkoholisches Lösungsmittel wie Methanol oder Ethanol verwendet. Die Kondensation wird im Allgemeinen in einem Temperaturbe- reich von +70°C bis +120°C durchgeführt [vgl. z.B. K. Lackey und D. D. Sternbach, Synthesis, 1993, 993-997; A. N. Boa et at., Bioorg. Med. Chem. 2005, 13 (6), 1945-1967].
Die Kondensationsreaktion zur Chinolin-4-carbonsäure der Formel (Vll-A) (Schema 2) erfolgt auf analoge Weise durch Erhitzen des o/f/?o-Aminophenylessigsäureesters der Formel (VIII) und des Diketons der Formel (IX) mit wässriger Säure, insbesondere konzentrierter Salzsäure. Als inertes Lösungsmittel für die Umsetzung wird auch hier bevorzugt Essigsäure eingesetzt.
Der o/f/70-Aminophenylessigsäureester der Formel (VIII) seinerseits kann in Anlehnung an ein in der Literatur beschriebenes Verfahren durch basenvermittelte Reaktion des a-Chloressigsäure- esters der Formel (XX) mit einem Nitrophenyl-Derivat der Formel (XXI) zum einem ortho- Nitrophenylessigsäureester der Formel (XXII) umgesetzt werden (Schema 3). Die nachfolgende Reduktion der Nitro-Gruppe zu einer Verbindung der Formel (XXI) kann beispielsweise durch ka- talytische Hydrierung erhalten werden [vgl. P. Beier ei a/., J. Org. Chem. 2011 , 76, 4781-4786].
Schema 3:
Die Verbindungen der Formel (lll-A) und (III-B) können in Abhängigkeit vom jeweiligen Substitutionsmuster beispielweise gemäß den in den nachfolgenden Schemata 4 bis 10 und im Experi- mentalteil bei den jeweiligen Beispielen beschriebenen Syntheserouten und in Analogie zu literaturbekannten Syntheseverfahren hergestellt werden:
Schema 4 (für Y = #1-(CH2)n-#2 mit n = 0, 1 , 2, 3 (lll-B-1 )).
(XIII) (XIV) (lll-B-1 )
[Alkyl = Methyl, Ethyl; Ar = subst. Phenyl, subst. Pyridyl; T = Ester-Schutzgruppe, z.B. Methyl, Ethyl, ie/f-Butyl; n = 0, 1 , 2, 3; a): für n=0: Di-ie/f-butyldicarbonat, LDA, -78 °C; für n=1 : ierf-Butyl- bromacetat, LDA oder LiHMDS, -78 °C; für n=2: ie/f-Butyl-3-brompropanoat, LDA oder LiHMDS, -78 °C; oder ie f-Butyl-acrylat, Kaliumcarbonat oder Natriumhydrid; für n=3: ie/f-Butyl-4- brombutanoat, LDA, -78 °C b): H , Raney-Nickel oder PtÜ2. LDA = Lithiumdiisopropylamid; LiHMDS = Lithium-bis(trimethylsilyl)amid].
Schema 5 (für R8 = Fluor (lll-B-2), Hydroxy (lll-B-3)).
(lll-B-3)
[Ar = subst. Phenyl, subst. Pyridyl; T = Ester-Schutzgruppe, z.B. Methyl, Ethyl, ie f-Butyl; n = 0, 1 , 2, 3; a): z.B. KCN oder TMSCN; b) DAST; c): H2, Katalysator, z.B. Raney-Nickel oder Pt02. DAST = Diethylaminoschwefeltrifluorid].
Schema 6 (für R
7A/R
7B = H, Me (lll-B-4)).
(XVII) 2 (XVIII) 2 (lll-B-4) [Ar = subst. Phenyl, subst. Pyridyl; T = Ester-Schutzgruppe, z.B. Methyl, Ethyl, ie/f-Butyl; a): Nit- roethan, Base, z.B. 2-ie/f-Butyl-1 ,1 ,3,3-tetramethylguanidin; b) Zinkstaub, Salzsäure].
Schema 7 (für R
10A/R
10B = Me, Me (lll-B-5) oder F, F (lll-B-6)).
(XXIII) (lll-B-6)
[Ar = subst. Phenyl, subst. Pyridyl; T = Ester-Schutzgruppe, z.B. Methyl, Ethyl, fe/f-Butyl; a): LDA, - 78 °C; b) Zink; c) H2, Raney-Nickel; d) H2, Pt02. LDA = Lithiumdiisopropylamid].
Schema 8 (für X = NH (lll-B-7) oder NMe (lll-B-8)).
(XXIV) (XXV) (XXVI) (lll-B-7/8)
[Ar = subst. Phenyl, subst. Pyridyl; T = Ester-Schutzgruppe, z.B. Methyl, Ethyl, fe/f-Butyl; a): TMS-CN; b) H2, Raney-Nickel].
Schema 9 (für X = S (lll-B-9) oder S02 (II l-B-10)).
(XIX) (XXVII) (lll-B-9) (lll-B-10) [Ar = subst. Phenyl, subst. Pyridyl; T = Ester-Schutzgruppe, z.B. Methyl, Ethyl, ie/f-Butyl; a) z.B. in DCM; b) SnCI2; c) Schützung Amino-Funktionalität, z.B. als NH-Boc: Di-ie f-butyldicarbonat, Triethylamin; d) mCPBA; e) Entschützung Amino-Funktionalität, z.B. HCI/Dioxan. Boc = tert- Butyloxycarbonyl; DCM = Dichlormethan].
Schema 10 (für X = O (lll-B-1 1 )).
(XVIII) (XXIX) (lll-B-11 )
[Ar = subst. Phenyl, subst. Pyridyl; T = Ester-Schutzgruppe, z.B. Methyl, Ethyl, ie/f-Butyl; PG = Schutzgruppe, z.B. Boc; a) Rh2(OAc)4; b) Entschützung Amino-Funktionalität, z.B. HCI/Dioxan. Boc = ie/f-Butyloxycarbonyl].
Eine Trennung der erfindungsgemäßen Verbindungen in die entsprechenden Enantiomere und/oder Diastereomere kann gegebenenfalls, je nach Zweckmäßigkeit, auch bereits auf der Stufe der Intermediate (lll-A), (III-B) oder (IV) erfolgen, welche dann in separierter Form gemäß der zuvor beschriebenen Reaktionssequenz weiter umgesetzt werden. Es kann zweckmäßig sein, die Amin-Funktionalität der Intermediate (lll-A) und (III-B) vor einer solchen Trennung mit einer Schutzgruppe, z.B. Boc, zu versehen und nach der Trennung anschließend wieder zu ent- schützen. Für eine solche Auftrennung der Stereoisomere von Intermediaten werden gleichfalls bevorzugt chromatographische Verfahren an achiralen bzw. chiralen Trennphasen angewandt.
Die Verbindungen der Formeln (V), (VI), (VIII), (IX), (X), (XI), (XIII), (XV), (XVII), (XIX), (XX), (XXII), (XXIV), (XXV), (XXVII), (XVIII) und (XXIX) sind gleichfalls kommerziell erhältlich oder als solche in der Literatur beschrieben, oder sie können, ausgehend von anderen kommerziell er- hältlichen Verbindungen, auf einfache Weise in Analogie zu literaturbekannten Verfahren hergestellt werden.
Detaillierte Vorschriften und weitere Literaturangaben befinden sich auch im Experimentellen Teil im Abschnitt zur Herstellung der Ausgangsverbindungen und Intermediate.
Weitere erfindungsgemäße Verbindungen der Formel (I) können, falls zweckmäßig, auch durch Umwandlungen von funktionellen Gruppen einzelner Reste und Substituenten, insbesondere den unter R1 und R5 aufgeführten, hergestellt werden, wobei von anderen, nach obigen Verfahren erhaltenen Verbindungen der Formel (I) oder deren Vorstufen ausgegangen wird. Diese Umwandlungen werden nach üblichen, dem Fachmann geläufigen Methoden durchgeführt und umfassen beispielsweise Reaktionen wie nukleophile oder elektrophile Substitutionsreaktionen, Übergangsmetall-vermittelte Kupplungsreaktionen, Herstellungs- und Additionsreaktionen von Metallorganylen (z.B. Grignard-Verbindungen oder Lithiumorganylen), Oxidations- und Reduktionsreaktionen, Hydrierung, Halogenierung (z.B. Fluorierung, Bromierung), Dehalogenierung, Aminierung, Alkylierung und Acylierung, die Bildung von Carbonsäureestern, Carbonsäureami- den und Sulfonamiden, die Esterspaltung und -hydrolyse sowie die Einführung und Entfernung temporärer Schutzgruppen.
Die erfindungsgemäßen Verbindungen besitzen wertvolle pharmakologische Eigenschaften und können zur Behandlung und/ oder Prophylaxe von Erkrankungen bei Menschen und Tieren verwendet werden.
Die erfindungsgemäßen Verbindungen stellen potente, chemisch und metabolisch stabile Antagonisten des FP-Rezeptors („FP-Antagonisten") dar und eignen sich daher zur Behandlung und/oder
Prävention von Erkrankungen und pathologischen Prozessen, insbesondere solcher, bei denen im Zuge eines Entzündungsgeschehens und/oder eines Gewebe- oder Gefäßumbaus der FP- Rezeptor involviert ist.
Dazu zählen im Sinne der vorliegenden Erfindung insbesondere Erkrankungen wie die Gruppe der interstitiellen idiopathischen Pneumonien, zu denen die idiopathische pulmonale Fibrose (IPF), die akute interstitielle Pneumonie, nicht-spezifische interstitielle Pneumonien, lymphoide interstitielle Pneumonien, respiratorische Bronchiolitis mit interstitieller Lungenerkrankung, kryptogene organisierende Pneumonien, desquamative interstitielle Pneumonien und nicht-klassifizierbare idiopathische interstitielle Pneumonien gehören, ferner granulomatöse interstitielle Lungenerkrankungen, rheumatoide Arthritis mit interstitieller Lungenerkrankung, interstitielle Lungenerkrankungen bekannter Ursache und andere interstitielle Lungenerkrankungen unbekannter Ursache, die pulmonale arterielle Hypertonie (PAH) und andere Formen der pulmonalen Hypertonie (PH), das Bronchiolitis obliterans-Syndrom (BOS), die chronisch-obstruktive Lungenerkrankung (COPD), Lun- gensarkoidose, das akute Atemwegssyndrom (ARDS), akute Lungenschädigung (ALI), alpha-1- Antitrypsin-Defizienz (AATD), Lungenemphysem (z.B. durch Zigarettenrauch induziertes Lungenemphysem), zystische Fibrose (CF), entzündliche und fibrotische Erkrankungen der Niere, chronische Darmentzündungen (IBD, Morbus Crohn, Colitis ulcerosa), Peritonitis, Peritonealfibrose, rheumatoide Erkrankungen, multiple Sklerose, entzündliche und fibrotische Hauterkrankungen, Sichelzellanämie sowie entzündliche und fibrotische Augenerkrankungen.
Die erfindungsgemäßen Verbindungen können weiterhin verwendet werden zur Behandlung und/ oder Prävention von asthmatischen Erkrankungen unterschiedlicher Schweregrade mit intermittierendem oder persistierendem Verlauf (refraktäres Asthma, bronchiales Asthma, allergisches Asthma, intrinsisches Asthma, extrinsisches Asthma, durch Medikamente oder durch Staub induziertes Asthma), von verschiedenen Formen der Bronchitis (chronische Bronchitis, infektiöse Bron- chitis, eosinophile Bronchitis), von Bronchiektasien, Pneumonie, Farmerlunge und verwandten Krankheiten, Husten- und Erkältungskrankheiten (chronischer entzündlicher Husten, iatrogener Husten), Nasenschleimhautentzündungen (einschließlich medikamentöse Rhinitis, vasomotorische Rhinitis und jahreszeitabhängige, allergische Rhinitis, z.B. Heuschnupfen) und von Polypen.
Die erfindungsgemäßen Verbindungen können darüber hinaus zur Behandlung und/oder Präven- tion von kardiovaskulären Erkrankungen eingesetzt werden, wie beispielsweise Bluthochdruck (Hypertonie), Herzinsuffizienz, koronare Herzerkrankung, stabile und instabile Angina pectoris, renale Hypertonie, periphere und kardiale Gefäßerkrankungen, Arrhythmien, Rhythmusstörungen der Vorhöfe und der Kammern sowie Überleitungsstörungen wie beispielsweise atrio-ventrikuläre Blockaden des Grades l-lll, supraventrikuläre Tachyarrhythmie, Vorhofflimmern, Vorhofflattern, Kammerflimmern, Kammerflattern, ventrikuläre Tachyarrhythmie, Torsade de pointes-Tachykardie, Extrasystolen des Vorhofs und des Ventrikels, AV-junktionale Extrasystolen, Sick-Sinus-Syndrom, Synkopen, AV-Knoten-Reentry-Tachykardie, Wolff-Parkinson-White-Syndrom, akutes Koronarsyn-
drom (ACS), autoimmune Herzerkrankungen (Perikarditis, Endokarditis, Valvolitis, Aortitis, Kardiomyopathien), Boxerkardiomyopathie, Aneurysmen, Schock wie kardiogener Schock, septischer Schock und anaphylaktischer Schock, ferner zur Behandlung und/oder Prävention von thrombo- embolischen Erkrankungen und Ischämien, wie myokardiale Ischämie, Myokardinfarkt, Hirnschlag, Herzhypertrophie, transistorische und ischämische Attacken, Präeklampsie, entzündliche kardiovaskuläre Erkrankungen, Spasmen der Koronararterien und peripherer Arterien, Ödembildung wie beispielsweise pulmonales Ödem, Hirnödem, renales Ödem oder Herzinsuffizienz-bedingtes Ödem, periphere Durchblutungsstörungen, Reperfusionsschäden, arterielle und venöse Thrombosen, Mikroalbuminurie, Herzmuskelschwäche, endotheliale Dysfunktion, mikro- und makro- vaskuläre Schädigungen (Vaskulitis), sowie zur Verhinderung von Restenosen beispielsweise nach Thrombolyse-Therapien, percutan-transluminalen Angioplastien (PTA), percutan-trans- luminalen Koronarangioplastien (PTCA), Herztransplantationen und Bypass-Operationen.
Im Sinne der vorliegenden Erfindung umfasst der Begriff Herzinsuffizienz sowohl akute als auch chronische Erscheinungsformen der Herzinsuffizienz wie auch spezifische oder verwandte Krankheitsformen hiervon, wie akute dekompensierte Herzinsuffizienz, Rechtsherzinsuffizienz, Linksherzinsuffizienz, Globalinsuffizienz, ischämische Kardiomyopathie, dilatative Kardiomyopathie, hypertrophe Kardiomyopathie, idiopathische Kardiomyopathie, diabetische Kardiomyopathie, angeborene Herzfehler, Herzklappenfehler, Herzinsuffizienz bei Herzklappenfehlern, Mitral- klappenstenose, Mitral klappen Insuffizienz, Aortenklappenstenose, Aortenklappeninsuffizienz, Trikuspidalstenose, Trikuspidalinsuffizienz, Pulmonalklappenstenose, Pulmonalklappeninsuffizi- enz, kombinierte Herzklappenfehler, Herzmuskelentzündung (Myokarditis), chronische Myokarditis, akute Myokarditis, virale Myokarditis, diabetische Herzinsuffizienz, alkoholtoxische Kardiomyopathie, kardiale Speichererkrankungen sowie diastolische und systolische Herzinsuffizienz.
Die erfindungsgemäßen Verbindungen eignen sich außerdem zur Behandlung und/oder Präven- tion von Nierenerkrankungen, insbesondere von Niereninsuffizienz und Nierenversagen. Im Sinne der vorliegenden Erfindung umfassen die Begriffe Niereninsuffizienz und Nierenversagen sowohl akute als auch chronische Erscheinungsformen hiervon wie auch diesen zugrundeliegende oder verwandte Nierenerkrankungen, wie renale Hypoperfusion, intradialytische Hypotonie, obstruktive Uropathie, Glomerulopathien, Glomerulonephritis, akute Glomerulonephritis, Glomerulosklerose, tubulointerstitielle Erkrankungen, nephropathische Erkrankungen wie primäre und angeborene Nierenerkrankung, Nierenentzündung, immunologische Nierenerkrankungen wie Nierentransplantat-Abstoßung und Immunkomplex-induzierte Nierenerkrankungen, durch toxische Substanzen induzierte Nephropathie, Kontrastmittel-induzierte Nephropathie, diabetische und nicht-diabetische Nephropathie, Pyelonephritis, Nierenzysten, Nephrosklerose, hypertensive Nephrosklerose und nephrotisches Syndrom, welche diagnostisch beispielsweise durch abnorm verminderte Kreatinin- und/oder Wasser-Ausscheidung, abnorm erhöhte Blutkonzentrationen von Harnstoff, Stickstoff, Kalium und/oder Kreatinin, veränderte Aktivität von Nierenenzymen wie
z.B. Glutamylsynthetase, veränderte Urinosmolarität oder Urinmenge, erhöhte Mikroalbuminurie, Makroalbuminurie, Läsionen an Glomerula und Arteriolen, tubuläre Dilatation, Hyperphosphatä- mie und/oder die Notwendigkeit zur Dialyse charakterisiert werden können. Die vorliegende Erfindung umfasst auch die Verwendung der erfindungsgemäßen Verbindungen zur Behandlung und/oder Prävention von Folgeerscheinungen einer Niereninsuffizienz, wie beispielsweise Hypertonie, Lungenödem, Herzinsuffizienz, Urämie, Anämie, Elektrolytstörungen (z.B. Hyperkalä- mie, Hyponaträmie) und Störungen im Knochen- und Kohlenhydrat-Metabolismus.
Darüber hinaus sind die erfindungsgemäßen Verbindungen zur Behandlung und/oder Prävention von Erkrankungen des Urogenitalsystems geeignet, wie beispielsweise benignes Prostata- Syndrom (BPS), benigne Prostatahyperplasie (BPH), benigne Prostatavergrößerung (BPE), Blasenentleerungsstörungen (BOO), untere Harnwegssyndrome (LUTS), neurogene überaktive Blase (OAB), Inkontinenz wie beispielsweise Misch-, Drang-, Stress- oder Überlauf-Inkontinenz (MUI, UUI, SUI, OUI), Beckenschmerzen sowie erektile Dysfunktion und weibliche sexuelle Dysfunktion.
Die erfindungsgemäßen Verbindungen können auch zur Behandlung von Erkrankungen des weiblichen Reproduktionssystems, wie Uterusmyome, Endometriose, Dysmenorrhöe und vorzeitige Geburtswehen, sowie periphär vermitteltem inflammatorischen Schmerz (z.B. bei symptomatischer Endometriose) verwendet werden. Weiterhin eignen sie sich zur Prophylaxe oder Behandlung von Hirsutismus und Hypertrichose.
Zudem besitzen die erfindungsgemäßen Verbindungen anti-inflammatorische Wirkung und können daher als entzündungshemmende Mittel zur Behandlung und/oder Prävention von Sepsis (SIRS), multiplem Organversagen (MODS, MOF), entzündlichen Erkrankungen der Niere, chronischen Darmentzündungen (IBD, Morbus Crohn, Colitis ulcerosa), Pankreatitis, Peritonitis, Cystitis, Urethritis, Prostatitis, Epidimytitis, Oophoritis, Salpingitis, Vulvovaginitis, rheumatoiden Erkrankungen, Arthrose, entzündlichen Erkrankungen des Zentralnervensystems, multipler Sklerose, entzünd- liehen Hauterkrankungen und entzündlichen Augenerkrankungen eingesetzt werden.
Die erfindungsgemäßen Verbindungen sind ferner zur Behandlung und/oder Prävention von fibro- tischen Erkrankungen der inneren Organe, wie beispielsweise der Lunge, des Herzens, der Niere, des Knochenmarks und insbesondere der Leber, sowie von dermatologischen Fibrosen und fibro- tischen Erkrankungen des Auges geeignet. Im Sinne der vorliegenden Erfindung umfasst der Be- griff fibrotische Erkrankungen insbesondere solche Erkrankungen wie Leberfibrose, Leberzirrhose, Lungenfibrose, Endomyokardfibrose, Nephropathie, Glomerulonephritis, interstitielle Nierenfibrose, fibrotische Schäden in Folge von Diabetes, Knochen marksfibrose, Peritonealfibrose und ähnliche fibrotische Erkrankungen, Sklerodermie, Morphaea, Keloide, hypertrophe Narbenbildung, Naevi, diabetische Retinopathie, proliferative Vitroretinopathie und Erkrankungen des Bindegewebes (z.B. Sarkoidose). Die erfindungsgemäßen Verbindungen können ebenso verwendet werden zur Förde-
rung der Wundheilung, zur Bekämpfung postoperativer Narbenbildung, z.B. nach Glaukom- Operationen, und zu kosmetischen Zwecken bei alternder oder verhornender Haut.
Auch können die erfindungsgemäßen Verbindungen zur Behandlung und/oder Prävention von Anämien verwendet werden, wie hämolytischen Anämien, insbesondere Hämoglobinopathien wie Sichelzellanämie und Thalassämien, megaloblastären Anämien, Eisenmangel-Anämien, Anämien durch akuten Blutverlust, Verdrängungsanämien und aplastischen Anämien.
Die erfindungsgemäßen Verbindungen sind zudem zur Behandlung von Krebserkrankungen geeignet, wie beispielsweise von Hautkrebs, Hirntumoren, Brustkrebs, Knochenmarktumoren, Leukämien, Liposarcomen, Karzinomen des Magen-Darm-Traktes, der Leber, Bauchspeicheldrüse, Lunge, Niere, Harnleiter, Prostata und des Genitaltraktes sowie von bösartigen Tumoren des lymphoproliferativen Systems, wie z.B. Hodgkin's und Non-Hodgkin's Lymphom.
Darüber hinaus können die erfindungsgemäßen Verbindungen eingesetzt werden zur Behandlung und/oder Prävention von Arteriosklerose, Lipidstoffwechselstörungen und Dyslipidämien (Hypolipo- proteinämie, Hypertriglyceridämie, Hyperlipidämie, kombinierte Hyperlipidämien, Hypercholesterol- ämie, Abetalipoproteinämie, Sitosterolämie), Xanthomatose, Tangier-Krankheit, Fettsucht (Adi- positas), Fettleibigkeit (Obesitas), metabolischen Erkrankungen (Metabolisches Syndrom, Hyper- glykämie, Insulin-abhängiger Diabetes, nicht-Insulin-abhängiger Diabetes, Gestationsdiabetes, Hyperinsulinämie, Insulinresistenz, Glukose-Intoleranz und diabetische Spätfolgen wie Retinopathie, Nephropathie und Neuropathie), von Erkrankungen des Gastrointestinaltrakts und des Ab- dornen (Glossitis, Gingivitis, Periodontitis, Oesophagitis, eosinophile Gastroenteritis, Mastocytose, Morbus Crohn, Colitis, Proctitis, Pruritis ani, Diarrhöe, Zöliakie, Hepatitis, Leberfibrose, Leberzirrhose, Pankreatitis und Cholecystitis), von Erkrankungen des Zentralen Nervensystems und von neurodegenerativen Störungen (Schlaganfall, Alzheimer'sche Krankheit, Parkinson'sche Krankheit, Demenz, Epilepsie, Depressionen, Multiple Sklerose), Immunerkrankungen, Schilddrüsen- erkrankungen (Hyperthyreose), Hauterkrankungen (Psoriasis, Akne, Ekzeme, Neurodermitis, vielfältige Formen der Dermatitis wie z.B. Dermatitis abacribus, Dermatitis actinica, Dermatitis allergica, Dermatitis ammoniacalis, Dermatitis artefacta, Dermatitis autogenica, Dermatitis atrophicans, Dermatitis calorica, Dermatitis combustionis, Dermatitis congelationis, Dermatitis cosmetica, Dermatitis escharotica, Dermatitis exfoliativa, Dermatitis gangraenose, Dermatitis haemostatica, Der- matitis herpetiformis, Dermatitis lichenoides, Dermatitis linearis, Dermatitis maligna, Dermatitis medimencatosa, Dermatitis palmaris et plantaris, Dermatitis parasitaria, Dermatitis photoallergica, Dermatitis phototoxica, Dermatitis pustularis, Dermatitis seborrhoica, Dermatitis solaris, Dermatitis toxica, Dermatitis ulcerosa, Dermatitis veneata, infektiöse Dermatitis, pyogene Dermatitis und Ro- sazea-artige Dermatitis, sowie Keratitis, Bullosis, Vasculitis, Cellulitis, Panniculitis, Lupus erythe- matodes, Erythema, Lymphome, Hautkrebs, Sweet-Syndrom, Weber-Christian-Syndrom, Narbenbildung, Warzenbildung, Frostbeulen), von entzündlichen Augenerkrankungen (Saccoidosis, Blepharitis, Conjunctivitis, Iritis, Uveitis, Chorioiditis, Ophthalmitis), viralen Erkrankungen (durch In-
fluenza-, Adeno- und Coronaviren, wie z.B. HPV, HCMV, HIV, SARS), von Erkrankungen des Skelettknochens und der Gelenke sowie der Skelettmuskel (vielfältige Formen der Arthritis wie z.B. Arthritis alcaptonurica, Arthritis ankylosans, Arthritis dysenterica, Arthritis exsudativa, Arthritis fun- gosa, Arthritis gonorrhoica, Arthritis mutilans, Arthritis psoriatica, Arthritis purulenta, Arthritis rheu- matica, Arthritis serosa, Arthritis syphilitica, Arthritis tuberculosa, Arthritis urica, Arthritis villonodula- ris pigmentosa, atypische Arthritis, hämophile Arthritis, juvenile chronische Arthritis, rheumatoide Arthritis und metastatische Arthritis, des weiteren das Still-Syndrom, Felty-Syndrom, Sjörgen- Syndrom, Clutton-Syndrom, Poncet-Syndrom, Pott-Syndrom und Reiter-Syndrom, vielfältige Formen der Arthropathien wie z.B. Arthropathie deformans, Arthropathie neuropathica, Arthropathie ovaripriva, Arthropathie psoriatica und Arthropathie tabica, systemische Sklerosen, vielfältige Formen der entzündlichen Myopathien wie z.B. Myopathie epidemica, Myopathie fibrosa, Myopathie myoglobinurica, Myopathie ossificans, Myopathie ossificans neurotica, Myopathie ossificans progressiva multiplex, Myopathie purulenta, Myopathie rheumatica, Myopathie trichinosa, Myopathie tropica und Myopathie typhosa, sowie das Günther-Syndrom und das Münchmeyer-Syndrom), von entzündlichen Arterienveränderungen (vielfältige Formen der Arteritis wie z.B. Endarteritis, Mesar- teritis, Periarteritis, Panarteritis, Arteritis rheumatica, Arteritis deformans, Arteritis temporalis, Arteritis cranialis, Arteritis gigantocellularis und Arteritis granulomatosa, sowie das Horton-Syndrom, Churg-Strauss-Syndrom und die Takayasu-Arteritis), des Muckle-Well-Syndroms, der Kikuchi- Krankheit, von Polychondritis, Sklerodermia sowie von weiteren Erkrankungen mit einer entzündli- chen oder immunologischen Komponente, wie beispielsweise Katarakt, Kachexie, Osteoporose, Gicht, Inkontinenz, Lepra, Sezary-Syndrom und paraneoplastisches Syndrom, bei Abstossungsre- aktionen nach Organtransplantationen und zur Wundheilung und Angiogenese insbesondere bei chronischen Wunden.
Aufgrund ihres biochemischen und pharmakologischen Eigenschaftsprofils eignen sich die erfin- dungsgemäßen Verbindungen insbesondere zur Behandlung und/oder Prävention von interstitiellen Lungenerkrankungen, vor allem der idiopathischen Lungenfibrose (IPF), sowie von pulmonaler Hypertonie (PH), Bronchiolitis obliterans-Syndrom (BOS), entzündlichen und fibrotischen Haut- und Augenerkrankungen und fibrotischen Erkrankungen der inneren Organe.
Die zuvor genannten, gut charakterisierten Krankheiten des Menschen können mit vergleich- barer Ätiologie auch in anderen Säugetieren vorkommen und dort ebenfalls mit den Verbindungen der vorliegenden Erfindung behandelt werden.
Im Sinne der vorliegenden Erfindung umfasst der Begriff "Behandlung" oder "behandeln" ein Hemmen, Verzögern, Aufhalten, Lindern, Abschwächen, Einschränken, Verringern, Unterdrücken, Zurückdrängen oder Heilen einer Krankheit, eines Leidens, einer Erkrankung, einer Verletzung oder einer gesundheitlichen Störung, der Entfaltung, des Verlaufs oder des Fortschreitens solcher Zustände und/oder der Symptome solcher Zustände. Der Begriff "Therapie" wird hierbei als synonym mit dem Begriff "Behandlung" verstanden.
Die Begriffe "Prävention", "Prophylaxe" oder "Vorbeugung" werden im Rahmen der vorliegenden Erfindung synonym verwendet und bezeichnen das Vermeiden oder Vermindern des Risikos, eine Krankheit, ein Leiden, eine Erkrankung, eine Verletzung oder eine gesundheitliche Störung, eine Entfaltung oder ein Fortschreiten solcher Zustände und/oder die Symptome solcher Zu- stände zu bekommen, zu erfahren, zu erleiden oder zu haben.
Die Behandlung oder die Prävention einer Krankheit, eines Leidens, einer Erkrankung, einer Verletzung oder einer gesundheitlichen Störung können teilweise oder vollständig erfolgen.
Weiterer Gegenstand der vorliegenden Erfindung ist somit die Verwendung der erfindungsgemäßen Verbindungen zur Behandlung und/oder Prävention von Erkrankungen, insbesondere der zuvor genannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Verbindungen zur Herstellung eines Arzneimittels zur Behandlung und/oder Prävention von Erkrankungen, insbesondere der zuvor genannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung ist ein Arzneimittel, enthaltend mindestens ei- ne der erfindungsgemäßen Verbindungen, zur Behandlung und/oder Prävention von Erkrankungen, insbesondere der zuvor genannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Verbindungen in einem Verfahren zur Behandlung und/oder Prävention von Erkrankungen, insbesondere der zuvor genannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung ist ein Verfahren zur Behandlung und/oder Prävention von Erkrankungen, insbesondere der zuvor genannten Erkrankungen, unter Verwendung einer wirksamen Menge von mindestens einer der erfindungsgemäßen Verbindungen.
Die erfindungsgemäßen Verbindungen können allein oder bei Bedarf in Kombination mit einer oder mehreren anderen pharmakologisch wirksamen Substanzen eingesetzt werden, solange diese Kombination nicht zu unerwünschten und inakzeptablen Nebenwirkungen führt. Weiterer Gegenstand der vorliegenden Erfindung sind daher Arzneimittel, enthaltend mindestens eine der erfindungsgemäßen Verbindungen und einen oder mehrere weitere Wirkstoffe, insbesondere zur Behandlung und/oder Prävention der zuvor genannten Erkrankungen. Als hierfür geeignete Kombinationswirkstoffe seien beispielhaft und vorzugsweise genannt:
· organische Nitrate und NO-Donatoren, wie beispielsweise Natriumnitroprussid, Nitroglycerin, Isosorbidmononitrat, Isosorbiddinitrat, Molsidomin oder SIN-1 , sowie inhalatives NO;
• Verbindungen, die den Abbau von cyclischem Guanosinmonophosphat (cGMP) und/oder cyc- lischem Adenosinmonophosphat (cAMP) inhibieren, wie beispielsweise Inhibitoren der Phosphodiesterasen (PDE) 1 , 2, 3, 4 und/oder 5, insbesondere PDE 5-lnhibitoren wie Sildenafil, Vardenafil, Tadalafil, Udenafil, Dasantafil, Avanafil, Mirodenafil oder Lodenafil;
• NO- und Häm-unabhängige Aktivatoren der löslichen Guanylatcyclase (sGC), wie insbesondere die in WO 01/19355, WO 01/19776, WO 01/19778, WO 01/19780, WO 02/070462 und WO 02/070510 beschriebenen Verbindungen;
• NO-unabhängige, jedoch Häm-abhängige Stimulatoren der löslichen Guanylatcyclase (sGC), wie insbesondere Riociguat sowie die in WO 00/06568, WO 00/06569, WO 02/42301 , WO
03/095451 , WO 2011/147809, WO 2012/004258, WO 2012/028647 und WO 2012/059549 beschriebenen Verbindungen;
• Prostacyclin-Analoga und IP-Rezeptor-Agonisten, wie beispielhaft und vorzugsweise lloprost, Beraprost, Treprostinil, Epoprostenol oder Selexipag;
· Endothelin-Rezeptor-Antagonisten, wie beispielhaft und vorzugsweise Bosentan, Darusentan, Ambrisentan oder Sitaxsentan;
• Verbindungen, die die humane neutrophile Elastase (HNE) inhibieren, wie beispielhaft und vorzugsweise Sivelestat oder DX-890 (Reltran);
• die Signaltransduktionskaskade inhibierende Verbindungen, beispielhaft und vorzugsweise aus der Gruppe der Kinase-Inhibitoren, insbesondere aus der Gruppe der Tyrosinkinase- und/oder Serin/Threoninkinase-Inhibitoren, wie beispielhaft und vorzugsweise Ninte- danib, Dasatinib, Nilotinib, Bosutinib, Regorafenib, Sorafenib, Sunitinib, Cediranib, Axitinib, Telatinib, Imatinib, Brivanib, Pazopanib, Vatalanib, Gefitinib, Erlotinib, Lapatinib, Canertinib, Lestaurtinib, Pelitinib, Semaxanib oder Tandutinib;
· Verbindungen, die den Ab- und Umbau der Extrazellulärmatrix inhibieren, beispielhaft und vorzugsweise Inhibitoren der Matrix-Metalloproteasen (MMPs), insbesondere Inhibitoren von Stromelysin, Kollagenasen, Gelatinasen und Aggrecanasen (hierbei vor allem von MMP-1 , MMP-3, MMP-8, MMP-9, MMP-10, MMP-1 1 und MMP-13) sowie der Metallo- Elastase (MMP-12);
· Verbindungen, die die Bindung von Serotonin an dessen Rezeptor blockieren, beispielhaft und vorzugsweise Antagonisten des 5-HT2B-Rezeptors wie PRX-08066;
• Antagonisten von Wachstumsfaktoren, Zytokinen und Chemokinen, beispielhaft und vorzugsweise Antagonisten von TGF-ß, CTGF, IL-1 , IL-4, IL-5, IL-6, IL-8, IL-13 und Integrinen;
• die Rho-Kinase inhibierende Verbindungen, wie beispielhaft und vorzugsweise Fasudil, Y-27632, SLx-21 19, BF-66851 , BF-66852, BF-66853, KI-23095 oder BA-1049;
• Verbindungen, die die lösliche Epoxidhydrolase (sEH) inhibieren, wie beispielsweise N,N'-D\- cyclohexylharnstoff, 12-(3-Adamantan-1-yl-ureido)-dodecansäure oder 1-Adamantan-1-yl-3- {5-[2-(2-eth oxyeth oxy)eth oxy] pen ty l}-h a rn stoff ;
• den Energiestoffwechsel des Herzens beeinflussende Verbindungen, wie beispielhaft und vor- zugsweise Etomoxir, Dichloracetat, Ranolazin oder Trimetazidin;
• anti-obstruktiv wirkende Mittel, wie sie z.B. zur Therapie der chronisch-obstruktiven Lungenerkrankung (COPD) oder eines Asthma bronchiale eingesetzt werden, beispielhaft und Vorzugs-
weise aus der Gruppe der inhalativ oder systemisch angewendeten ß-adrenergen Rezeptor- Agonisten (ß-Mimetika) und der inhalativ angewendeten anti-muscarinergen Substanzen;
• entzündungshemmende, immunmodulierende, immunsuppressive und/oder zytotoxische Mittel, beispielhaft und vorzugsweise aus der Gruppe der systemisch oder inhalativ angewende- ten Corticosteroide sowie Acetylcystein, Montelukast, Azathioprin, Cyclophosphamid, Hydro- xycarbamid, Azithromycin, Pirfenidon oder Etanercept;
• antifibrotisch wirkende Mittel, wie beispielhaft und vorzugsweise der Multikinase Inhibitor Nin- tedanib, Adenosin-A2b-Rezeptor-Antagonisten, Sphingosin-1-phosphat-Rezeptor 3 (S1 P3)- Antagonisten, Autotaxin-Inhibitoren, Lysophosphatidsäure-Rezeptor 1 (LPA-1 )- und Lyso- phosphatidsäure-Rezeptor 2 (LPA-2)-Antagonisten, Lysyloxidase (LOX)-lnhibitoren, Lysyloxi- dase-like-2-lnhibitoren, CTGF-Inhibitoren, IL-4-Antagonisten, IL-13-Antagonisten, avß6-lnte- grin-Antagonisten, TGF-ß-Antagonisten, Inhibitoren des Wnt-Signalwegs oder CCR2- Antagonisten;
• antithrombotisch wirkende Mittel, beispielhaft und vorzugsweise aus der Gruppe der Throm- bozytenaggregationshemmer, der Antikoagulantien und der profibrinolytischen Substanzen;
• den Blutdruck senkende Wirkstoffe, beispielhaft und vorzugsweise aus der Gruppe der Calcium-Antagonisten, Angiotensin All-Antagonisten, ACE-Hemmer, Vasopeptidase-Inhibitoren, Endothelin-Antagonisten, Renin-Inhibitoren, α-Rezeptoren-Blocker, ß-Rezeptoren-Blocker, Mineralocorticoid-Rezeptor-Antagonisten sowie der Diuretika;
· den Fettstoffwechsel verändernde Wirkstoffe, beispielhaft und vorzugsweise aus der Gruppe der Thyroidrezeptor-Agonisten, Cholesterinsynthese-Inhibitoren wie beispielhaft und vorzugsweise HMG-CoA-Reduktase- oder Squalensynthese-Inhibitoren, der ACAT-Inhibitoren, CETP- Inhibitoren, MTP-Inhibitoren, PPAR-a-, PPAR-γ- und/oder PPAR-8-Agonisten, Cholesterin- Absorptionshemmer, Lipase-Inhibitoren, polymeren Gallensäureadsorber, Gallensäure-Reab- sorptionshemmer und Lipoprotein(a)-Antagonisten; und/oder
• Chemotherapeutika, wie sie z.B. zur Therapie von Neubildungen (Neoplasien) der Lunge oder anderer Organe eingesetzt werden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem ß-adrenergen Rezeptor-Agonisten, wie beispielhaft und vor- zugsweise Albuterol, Isoproterenol, Metaproterenol, Terbutalin, Fenoterol, Formoterol, Repro- terol, Salbutamol oder Salmeterol, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einer anti-muscarinergen Substanz, wie beispielhaft und vorzugsweise Ipratropiumbromid, Tiotropiumbromid oder Oxitropiumbromid, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Corticosteroid, wie beispielhaft und vorzugsweise Prednison,
Prednisolon, Methylprednisolon, Triamcinolon, Dexamethason, Beclomethason, Betamethason, Flunisolid, Budesonid oder Fluticason, verabreicht.
Unter antithrombotisch wirkenden Mittel werden vorzugsweise Verbindungen aus der Gruppe der Thrombozytenaggregationshemmer, der Antikoagulantien und der profibrinolytischen Sub- stanzen verstanden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Thrombozytenaggregationshemmer, wie beispielhaft und vorzugsweise Aspirin, Clopidogrel, Ticlopidin oder Dipyridamol, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem Thrombin-Inhibitor, wie beispielhaft und vorzugsweise Ximela- gatran, Melagatran, Dabigatran, Bivalirudin oder Clexane, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem GPIIb/llla-Antagonisten, wie beispielhaft und vorzugsweise Tirofiban oder Abciximab, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Faktor Xa-Inhibitor, wie beispielhaft und vorzugsweise Riva- roxaban, Apixaban, Fidexaban, Razaxaban, Fondaparinux, Idraparinux, DU-176b, PMD-31 12, YM-150, KFA-1982, EMD-503982, MCM-17, MLN-1021 , DX 9065a, DPC 906, JTV 803, SSR- 126512 oder SSR-128428, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit Heparin oder einem low molecular weight (LMW)-Heparin-Derivat verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Vitamin K-Antagonisten, wie beispielhaft und vorzugsweise Coumarin, verabreicht.
Unter den Blutdruck senkenden Mitteln werden vorzugsweise Verbindungen aus der Gruppe der Calcium-Antagonisten, Angiotensin All-Antagonisten, ACE-Hemmer, Endothelin-Antagonisten, Renin-Inhibitoren, α-Rezeptoren-Blocker, ß-Rezeptoren-Blocker, Mineralocorticoid-Rezeptor- Antagonisten sowie der Diuretika verstanden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Calcium-Antagonisten, wie beispielhaft und vorzugsweise Nifedipin, Amlodipin, Verapamil oder Diltiazem, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem αι-Rezeptoren-Blocker, wie beispielhaft und vorzugsweise Prazosin, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem ß-Rezeptoren-Blocker, wie beispielhaft und vorzugsweise Propranolol, Atenolol, Timolol, Pindolol, Alprenolol, Oxprenolol, Penbutolol, Bupranolol, Metipranolol, Nadolol, Mepindolol, Carazalol, Sotalol, Metoprolol, Betaxolol, Celiprolol, Bisoprolol, Carteolol, Esmolol, Labetalol, Carvedilol, Adaprolol, Landiolol, Nebivolol, Epanolol oder Bucindolol, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem Angiotensin All-Antagonisten, wie beispielhaft und vorzugsweise Losartan, Candesartan, Valsartan, Telmisartan oder Embursatan, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem ACE-Hemmer, wie beispielhaft und vorzugsweise Enalapril, Captopril, Lisinopril, Ramipril, Delapril, Fosinopril, Quinopril, Perindopril oder Trandopril, verab- reicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Endothelin-Antagonisten, wie beispielhaft und vorzugsweise Bosentan, Darusentan, Ambrisentan oder Sitaxsentan, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem Renin-Inhibitor, wie beispielhaft und vorzugsweise Aliskiren, SPP-600 oder SPP-800, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Mineralocorticoid-Rezeptor-Antagonisten, wie beispielhaft und vorzugsweise Spironolacton, Eplerenon oder Finerenon, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Diuretikum, wie beispielhaft und vorzugsweise Furosemid, Bumetanid, Torsemid, Bendroflumethiazid, Chlorthiazid, Hydrochlorthiazid, Hydroflumethiazid, Methyclothiazid, Polythiazid, Trichlormethiazid, Chlorthalidon, Indapamid, Metolazon, Quineth- azon, Acetazolamid, Dichlorphenamid, Methazolamid, Glycerin, Isosorbid, Mannitol, Amilorid o- der Triamteren, verabreicht.
Unter den Fettstoffwechsel verändernden Mitteln werden vorzugsweise Verbindungen aus der Gruppe der CETP-Inhibitoren, Thyroidrezeptor-Agonisten, Cholesterinsynthese-Inhibitoren wie HMG-CoA-Reduktase- oder Squalensynthese-Inhibitoren, der ACAT-Inhibitoren, MTP-Inhibi- toren, PPAR-a-, PPAR-γ- und/oder PPAR-8-Agonisten, Cholesterin-Absorptionshemmer, poly-
meren Gallensäureadsorber, Gallensäure-Reabsorptionshemmer, Lipase-Inhibitoren sowie der Lipoprotein(a)-Antagonisten verstanden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem CETP-Inhibitor, wie beispielhaft und vorzugsweise Torcetrapib (CP-529 414), JJT-705 oder CETP-vaccine (Avant), verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Thyroidrezeptor-Agonisten, wie beispielhaft und vorzugsweise D-Thyroxin, 3,5,3'-Triiodothyronin (T3), CGS 23425 oder Axitirome (CGS 26214), verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem HMG-CoA-Reduktase-lnhibitor aus der Klasse der Statine, wie beispielhaft und vorzugsweise Lovastatin, Simvastatin, Pravastatin, Fluvastatin, Atorvastatin, Rosuvastatin oder Pitavastatin, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Squalensynthese-Inhibitor, wie beispielhaft und vorzugsweise BMS-188494 oder TAK-475, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem ACAT-Inhibitor, wie beispielhaft und vorzugsweise Avasimibe, Melinamide, Pactimibe, Eflucimibe oder SMP-797, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem MTP-Inhibitor, wie beispielhaft und vorzugsweise Implitapide, BMS-201038, R-103757 oder JTT-130, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem PPAR-y-Agonisten, wie beispielhaft und vorzugsweise Pio- glitazone oder Rosiglitazone, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem PPAR-8-Agonisten, wie beispielhaft und vorzugsweise GW 501516 oder BAY 68-5042, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Cholesterin-Absorptionshemmer, wie beispielhaft und vor- zugsweise Ezetimibe, Tiqueside oder Pamaqueside, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Lipase-Inhibitor, wie beispielhaft und vorzugsweise Orlistat, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem polymeren Gallensäureadsorber, wie beispielhaft und vorzugsweise Cholestyramin, Colestipol, Colesolvam, CholestaGel oder Colestimid, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem Gallensäure-Reabsorptionshemmer, wie beispielhaft und vorzugsweise AS BT (= IBAT)-lnhibitoren wie z.B. AZD-7806, S-8921 , AK-105, BARI-1741 , SC-435 oder SC-635, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Lipoprotein(a)-Antagonisten, wie beispielhaft und vorzugs- weise Gemcabene calcium (CI-1027) oder Nicotinsäure, verabreicht.
Besonders bevorzugt sind Kombinationen der erfindungsgemäßen Verbindungen mit einem oder mehreren weiteren Wirkstoffen ausgewählt aus der Gruppe bestehend aus PDE 5-lnhibitoren, sGC-Aktivatoren, sGC-Stimulatoren, Prostacyclin-Analoga, IP-Rezeptor-Agonisten, Endothelin- Antagonisten, die Signaltransduktionskaskade inhibierenden Verbindungen und Pirfenidon. Weiterer Gegenstand der vorliegenden Erfindung sind Arzneimittel, die mindestens eine erfindungsgemäße Verbindung, üblicherweise zusammen mit einem oder mehreren inerten, nichttoxischen, pharmazeutisch geeigneten Hilfsstoffen enthalten, sowie deren Verwendung zu den zuvor genannten Zwecken.
Die erfindungsgemäßen Verbindungen können systemisch und/oder lokal wirken. Zu diesem Zweck können sie auf geeignete Weise appliziert werden, wie z.B. oral, parenteral, pulmonal, nasal, sublingual, lingual, buccal, rectal, vaginal, dermal, transdermal, conjunctival, otisch oder als Implantat bzw. Stent.
Für diese Applikationswege können die erfindungsgemäßen Verbindungen in geeigneten Applikationsformen verabreicht werden.
Für die orale Applikation eignen sich nach dem Stand der Technik funktionierende schnell und/oder modifiziert die erfindungsgemäßen Verbindungen abgebende Applikationsformen, die die erfindungsgemäßen Verbindungen in kristalliner und/ oder amorphisierter und/oder gelöster Form enthalten, wie z.B. Tabletten (nichtüberzogene oder überzogene Tabletten, beispielsweise mit magensaftresistenten oder sich verzögert auflösenden oder unlöslichen Überzügen, die die Freisetzung der erfindungsgemäßen Verbindung kontrollieren), in der Mundhöhle schnell zerfallende Tabletten oder Filme/Oblaten, Filme/Lyophylisate, Kapseln (beispielsweise Hart- oder Weichgelatinekapseln), Dragees, Granulate, Pellets, Pulver, Emulsionen, Suspensionen, Aerosole oder Lösungen.
Die parenterale Applikation kann unter Umgehung eines Resorptionsschrittes geschehen (z.B. in- travenös, intraarteriell, intrakardial, intraspinal oder intralumbal) oder unter Einschaltung einer Re-
Sorption (z.B. intramuskulär, subcutan, intracutan, percutan, intravitreal oder intraperitoneal). Für die parenterale Applikation eignen sich als Applikationsformen u.a. Injektions- und Infusionszubereitungen in Form von Lösungen, Suspensionen, Emulsionen, Lyophilisaten oder sterilen Pulvern.
Für die sonstigen Applikationswege eignen sich z.B. Inhalationsarzneiformen (u.a. Pulverinhala- toren, Nebulizer), Nasentropfen, -lösungen, -sprays; lingual, sublingual oder buccal zu applizierende Tabletten, Filme/Oblaten oder Kapseln, Suppositorien, Augentropfen, Augensalben, Augenbäder, okulare Inserte, Ohrentropfen, -sprays, -pulver, -Spülungen, -tampons, , Vaginalkapseln, wässrige Suspensionen (Lotionen, Schüttelmixturen), lipophile Suspensionen, Emulsionen, Mikroemulsionen, Salben, Cremes, transdermale therapeutische Systeme (wie beispielsweise Pflaster), Milch, Pasten, Schäume, Streupuder, Implantate oder Stents.
Bevorzugt sind die orale und die parenterale Applikation, insbesondere die orale, die intravenöse und die intrapulmonale (inhalative) Applikation.
Die erfindungsgemäßen Verbindungen können in die angeführten Applikationsformen überführt werden. Dies kann in an sich bekannter Weise durch Mischen mit pharmazeutisch geeigneten Hilfsstoffen geschehen. Zu diesen Hilfsstoffen zählen u.a.
• Füll- und Trägerstoffe (beispielsweise Cellulose, mikrokristalline Cellulose wie z.B. Avi- cel®, Laktose, Mannitol, Stärke, Calciumphosphate wie z.B. Di-Cafos®),
• Salbengrundlagen (beispielsweise Vaselin, Paraffine, Triglyceride, Wachse, Wollwachs, Wollwachsalkohole, Lanolin, hydrophile Salbe, Polyethylenglycole),
· Suppositoriengrundlagen (zum Beispiel Polyethylenglycole, Kakaobutter, Hartfett),
• Lösungsmittel (z.B. Wasser, Ethanol, Isopropanol, Glycerol, Propylenglycol, mittelkettige Triglyceride fette Öle, flüssige Polyethylenglycole, Paraffine),
• Tenside, Emulgatoren, Dispergier- oder Netzmittel (beispielsweise Natriumdodecylsulfat, Lecithin, Phospholipide, Fettalkohole wie z.B. Lanette®, Sorbitanfettsäureester wie z.B. Span®, Polyoxy-ethylen-Sorbitanfettsäureester wie z.B. Tween®, Polyoxyethylen-
Fettsäureglyceride wie z.B. Cremophor®, Polyoxethlyen-Fettsäureester, Polyoxethlyen- Fettalkoholether, Glycerolfettsäureester, Poloxamere wie z.B. Pluronic®),
• Puffersubstanzen sowie Säuren und Basen (beispielsweise Phosphate, Carbonate, Cit- ronensäure, Essigsäure, Salzsäure, Natronlauge, Ammoniumcarbonat, Trometamol, Triethanolamin),
• Isotonisierungsmittel (beispielsweise Glucose, Natriumchlorid),
• Adsorptionsmittel (beispielsweise hochdisperse Siliciumdioxide),
• Viskositätserhöhende Mittel, Gelbildner, Verdickungs- bzw. Bindemittel (beispielsweise Polyvinylpyrrolidon, Methylcellulose, Hydroxypropylmethylcellulose, Hydroxypropyl- cellulose, Carboxymethylcellulose-Natrium, Stärke, Carbomere, Polyacrylsäuren wie z.B.
Carbopol®, Alginate, Gelatine),
• Sprengmittel (beispielsweise modifizierte Stärke, Carboxymethylcellulose-Natrium, Natri- umstärkeglycolat wie z.B. Explotab®, quervernetztes Polyvinylpyrrolidon, Croscarmello- se-Natrium wie z.B. AcDiSol®),
• Fließregulier-, Schmier-, Gleit- und Formtrenn mittel (beispielsweise Magnesiumstearat, Stearinsäure, Talkum, hochdisperse Siliciumdioxide wie z.B. Aerosil®),
• Überzugsmittel (beispielsweise Zucker, Schellac) sowie Filmbildemittel für sich schnell oder modifiziert auflösende Filme bzw. Diffusionsmembranen (beispielsweise Polyvinylpyrro- lidone wie z.B. Kollidon®, Polyvinylalkohol, Hydroxypropylmethylcellulose, Hydroxypropyl- cellulose, Ethylcellulose, Hydroxypropylmethylcellulosephthalat, Celluloseacetat, Cellulose- acetatphthtalat, Polyacrylate, Polymethacrylate wie z.B. Eudragit®),
• Kapselmaterialien (z.B. Gelatine, Hydroxypropylmethylcellulose),
• Synthetische Polymere (beispielsweise Polylactide, Polyglycolide, Polyacrylate, Polymethacrylate wie z.B Eudragit®, Polyvinylpyrrolidone wie z.B. Kollidon®, Polyvinylalcohole, Polyvinylacetate, Polyethylenoxide, Polyethylenglycole und deren Copolymere und Blockcopolymere),
• Weichmacher (beispielsweise Polyethylenglycole, Propylenglykol, Glycerol, Triacetin, Triacetylcitrat, Dibutylphthalat),
• Penetrationsenhancer,
• Stabilisatoren (z.B. Antioxidantien wie beispielsweise Ascorbinsäure, Ascorbylpalmitat, Natriumascorbat, Butylhydroxyanisol, Butylhydroxytoluol, Propylgallat),
• Konservierungsmittel (beispielsweise Parabene, Sorbinsäure, Thiomersal, Benzalkoni- umchlorid, Chlorhexidinacetat, Natriumbenzoat),
• Farbstoffe (z.B. anorganische Pigmente wie beispielsweise Eisenoxide, Titandioxid),
• Aromen, Süßungsmittel, Geschmacks- und / oder Geruchskorrigentien.
Im Allgemeinen hat es sich als vorteilhaft erwiesen, bei parenteraler Applikation Mengen von etwa 0.001 bis 1 mg/kg, vorzugsweise etwa 0.01 bis 0.5 mg/kg Körpergewicht zur Erzielung wirksamer Ergebnisse zu verabreichen. Bei oraler Applikation beträgt die Dosierung etwa 0.01 bis 100 mg/kg, vorzugsweise etwa 0.01 bis 20 mg/kg und ganz besonders bevorzugt 0.1 bis 10 mg/kg Körpergewicht. Bei intrapulmonaler Applikation beträgt die Menge im Allgemeinen etwa 0.1 bis 50 mg je Inhalation.
Trotzdem kann es gegebenenfalls erforderlich sein, von den genannten Mengen abzuweichen, und zwar in Abhängigkeit von Körpergewicht, Applikationsweg, individuellem Verhalten gegenüber dem Wirkstoff, Art der Zubereitung und Zeitpunkt bzw. Intervall, zu welchem die Applikation erfolgt. So kann es in einigen Fällen ausreichend sein, mit weniger als der vorgenannten Min- destmenge auszukommen, während in anderen Fällen die genannte obere Grenze überschritten
werden muss. Im Falle der Applikation größerer Mengen kann es empfehlenswert sein, diese in mehreren Einzelgaben über den Tag zu verteilen.
Weiterer Gegenstand der vorliegenden Erfindung sind pharmazeutische Zusammensetzungen, die mindestens eine erfindungsgemäße Verbindung, üblicherweise zusammen mit einem oder mehreren pharmazeutisch geeigneten Hilfsstoffen enthalten, sowie deren Verwendung gemäß der vorliegenden Erfindung.
Die nachfolgenden Ausführungsbeispiele erläutern die Erfindung. Die Erfindung ist nicht auf die Beispiele beschränkt.
A. Beispiele Abkürzungen und Akronyme:
[α]ϋ20 spezifischer Drehwinkel (bei Polarometrie)
br. breit (bei NMR)
ca. circa
d Dublett (bei NMR)
dd Dublett von Dublett (bei NMR)
DIPEA Λ/,/V-Diisopropylethylamin
DMF Λ/,/V-Dimethylformamid
DMSO Dimethylsulfoxid
ΔΤ Erwärmung, Temperaturerhöhung (in Reaktionsschemata)
d. Th. der Theorie (bei chemischer Ausbeute)
ee Enantiomerenüberschuss
El Elektronenstoß-Ionisation (bei MS)
ESI Elektrospray-Ionisation (bei MS)
Et Ethyl
h Stunde(n)
HATU 0-(7-Azabenzotriazol-1-yl)-/V,/V,/V',/V'-tetramethyluronium-hexafluorophosphat
(CAS RN 148893-10-1 )
HPLC Hochdruck-, Hochleistungsflüssigchromatographie
IPr Isopropyl
LC Flüssigchromatographie
LC-MS Flüssigchromatographie-gekoppelte Massenspektrometrie
LDA Lithiumdiisopropylamid
LiHMDS Lithium-bis(trimethylsilyl)amid
m Multiple« (bei NMR)
M Molar
Me Methyl
min Minute(n)
MS Massenspektrometrie
NMR Kernresonanzspektrometrie
q Quartett (bei NMR)
qd Quartett von Dublett (bei NMR)
RP reverse phase (Umkehrphase, bei HPLC)
RT Raumtemperatur
Rt Retentionszeit (bei HPLC, LC/MS)
s Singulett (bei NMR)
SFC superkritische Flüssigkeitschromatographie (supercritical fluid chromatography) t Triplett (bei NMR)
td Triplett von Dublett (bei NMR)
TFA Trifluoressigsäure
tert tertiär
THF Tetrahydrofuran
TosMIC para-Toluolsulfonylmethylisocyanid
UPLC Ultra Performance Liquid Chromatography
UV Ultraviolett-Spektrometrie
Andere Abkürzungen haben ihre dem Fachmann an sich geläufigen Bedeutungen.
HPLC- und LC/MS-Methoden:
Methode 1 (LC-MS):
Gerätetyp MS: Thermo Scientific FT-MS; Gerätetyp UHPLC+: Thermo Scientific UltiMate 3000; Säule: Waters, HSST3, 2.1 x 75 mm, C18 1 .8 μηι; Eluent A: 1 I Wasser + 0.01 % Ameisensäure; Eluent B: 1 I Acetonitril + 0.01 % Ameisensäure; Gradient: 0.0 min 10% B -> 2.5 min 95% B -> 3.5 min 95% B; Ofen: 50°C; Fluss: 0.90 ml/min; UV-Detektion: 210 nm/ Optimum Integration Path 210-300 nm
Methode 2 (LC-MS):
Instrument: Waters ACQUITY SQD UPLC System; Säule: Waters Acquity UPLC HSS T3 1 .8 μηι 50 x 1 mm; Eluent A: 1 I Wasser + 0.25 ml 99%ige Ameisensäure , Eluent B: 1 I Acetonitril + 0.25 ml 99%ige Ameisensäure; Gradient: 0.0 min 90% A -> 1 .2 min 5% A -> 2.0 min 5% A Ofen: 50°C; Fluss: 0.40 ml/min; UV-Detektion: 208 - 400 nm.
Methode 3 (LC-MS):
Instrument: Waters ACQUITY SQD UPLC System; Säule: Waters Acquity UPLC HSS T3 1 .8 μηι 50 x 1 mm; Eluent A: 1 I Wasser + 0.25 ml 99%ige Ameisensäure , Eluent B: 1 I Acetonitril +
0.25 ml 99%ige Ameisensäure; Gradient: 0.0 min 95% A -> 6.0 min 5% A -> 7.5 min 5% A Ofen: 50°C; Fluss: 0.35 ml/min; UV-Detektion: 210 - 400 nm.
Methode 4 (LC-MS):
Instrument: Waters Single Quad MS System; Instrument Waters UPLC Acquity; Säule : Waters BEH C18 1.7 m 50 x 2.1 mm; Eluent A: 1 I Wasser + 1.0 ml_ (25%ig Ammoniak)/L, Eluent B: 1 I Acetonitril; Gradient: 0.0 min 92% A -> 0.1 min 92% A -> 1.8 min 5% A -> 3.5 min 5% A; Ofen: 50°C; Fluss: 0.45 mL/min; UV-Detektion: 210 nm (208-400 nm)
Methode 5 (LC-MS):
Instrument: Waters Acquity UPLCMS SingleQuad; Säule: Acquity UPLC BEH C18 1.7 μηι, 50x2.1 mm; Eluent A: Wasser + 0.1 Vol-% Ameisensäure (99%), Eluent B: Acetonitril; Gradient: 0.0 min 1 % B -> 1.6 min 99% B -> 2.0 min 99% B; Fluss 0.8 ml/min; Temperatur: 60 °C; DAD scan: 210-400 nm.
Methode 6 (LC-MS):
Instrument: Waters Acquity UPLCMS SingleQuad; Säule: Acquity UPLC BEH C18 1.7 μηι, 50x2.1 mm; Eluent A: Wasser + 0.2 Vol-% wässriger Ammoniak (32%), Eluent B: Acetonitril; Gradient: 0.0 min 1 % B — > 1.6 min 99% B -> 2.0 min 99% B; Fluss 0.8 ml/min; Temperatur: 60 °C; DAD scan: 210-400 nm.
Methode 7 (LC-MS):
Instrument: Agilent MS Quad 6150;HPLC: Agilent 1290; Säule: Waters Acquity UPLC HSS T3 1.8 m 50 x 2.1 mm; Eluent A: 1 I Wasser + 0.25 ml 99%ige Ameisensäure , Eluent B: 1 I
Acetonitril + 0.25 ml 99%ige Ameisensäure; Gradient: 0.0 min 90% A -> 0.3 min 90% A -> 1.7 min 5% A -> 3.0 min 5% A Ofen: 50°C; Fluss: 1 ,20 ml/min; UV-Detektion: 205 - 305 nm.
Methode 8 (LC-MS):
Gerätetyp MS: ThermoFisherScientific LTQ-Orbitrap-XL; Gerätetyp HPLC: Agilent 1200SL; Säule: Agilent, POROSHELL 120, 3 x 150 mm, SB - C18 2.7 m; Eluent A: 1 I Wasser + 0.1 % Trifluoressigsäure; Eluent B: 1 I Acetonitril + 0.1 % Trifluoressigsäure; Gradient: 0.0 min 2% B— > 0.3 min 2% B -> 5.0 min 95% B -> 10.0 min 95% B; Ofen: 40°C; Fluss: 0.75 ml/min; UV- Detektion: 210 nm
Methode 9 (LC-MS):
Detektion: MSD ESI pos/neg; Säule: Waters XSelect CSH C18, 30 x 2.1 mm, 3.5 Mm; Eluent A: 0.1% Ameisensäure in Acetonitril; Eluent B: 0.1% Ameisensäure in Wasser; Gradient: 0.0 min 5% A -> 1.6 min 98% A -> 3.0 min 98% A; Ofen: 35°C; Fluss: 1.0 ml/min; UV-Detektion: 220-320 nm
Methode 10 (LC-MS):
Detektion: MSD ES pos; Säule: Waters XSelect CSH C18, 30 x 2.1 mm, 3.5 [Jim; Eluent A: 95% Acetonitril + 5% 10 mM Ammoniumhydrogencarbonat in Wasser; Eluent B: 10 mM Ammonium- hydrogencarbonat in Wasser; Gradient: 0.0 min 5% A -> 1 .6 min 98% A -> 3.0 min 98% A; Ofen: 25°C; Fluss: 1 .0 ml/min; UV-Detektion: 220 nm
Methode 1 1 (LC-MS):
Detektion: MSD ESI pos/neg; Säule: Waters XSelect CSH C18, 50 x 2.1 mm, 3.5 μηι; Eluent A: 0.1 % Ameisensäure in Acetonitril; Eluent B: 0.1 % Ameisensäure in Wasser; Gradient: 0.0 min 5% A -> 3.5 min 98% A -> 6.0 min 98% A; Ofen: 35°C; Fluss: 0.8 ml/min; UV-Detektion: 270 nm Methode 12 (GC-MS):
Instrument: Thermo Scientific DSQII , Thermo Scientific Trace GC Ultra; Säule: Restek RTX- 35MS, 15 m x 200 m x 0.33 [Jim; konstanter Fluss mit Helium: 1 .20 ml/min; Ofen: 60°C; Inlet: 220°C; Gradient: 60°C, 30°C/min -> 300°C (3.33 min halten).
Methode 13 (GC-MS):
Instrument: Agilent 6890N; Detektion: MSD El pos; Säule: RXi-5MS 20 m, I D 180 [Jim; df 0.18 [ m; Detektionstemperatur: 280 °C; Massenbereich 50-550; Träger-Gas: Helium; durchschnittliche Geschwindigkeit: 50 cm/s; Initiale Temperatur: 100 °C, initiale Zeit: 1 .5 min; Solvens-Delay: 1 .0 min; finale Temperatur: 250 °C, finale Zeit: 250 °C; Injektionsvolumen 1 μΙ; Slit-Ratio 100:1 .
Methode 14 (präparative HPLC):
Säule: Chromatorex C18, 10 μηι, 250 x 40 mm; Eluent A: Wasser , Eluent B: Acetonitril; Injektion bei 3 min; Gradient: 0.0 min 30% B -> 6.0 min 30% B -> 27 min 95% B -> 38 min 95% B -> 39 min 30% B -> 40.2 min 30% B; Fluss: 50 ml/min. UV-Detektion: 210 nm
Methode 15 (präparative HPLC):
Säule: Chromatorex C18, 10 [Jim, 125 x 30 mm; Eluent A: Wasser + 0.1 % TFA, Eluent B: Acetonitril; Injektion bei 3 min; Gradient: 0.0 min 30% B -> 5.5 min 30% B -> 17.65 min 95% B -> 19.48 min 95% B -> 19.66 min 30% B -> 20.51 min 30% B; Fluss: 75 ml/min. UV-Detektion: 210 nm
Methode 16 (präparative HPLC):
Säule: Chromatorex C18, 10 [Jim, 250 x 40 mm; Eluent A: Wasser + 0.1 % TFA, Eluent B: Acetonitril; Injektion bei 3 min; Gradient: 0.0 min 30% B -> 6.0 min 30% B -> 27 min 95% B -> 38 min 95% B -> 39 min 30% B -> 40.2 min 30% B; Fluss: 50 ml/min. UV-Detektion: 210 nm
Methode 17 (präparative HPLC):
Säule: Chromatorex C18, 10 μηη, 125 x 30 mm; Eluent A: Wasser, Eluent B: Acetonitril; Injektion bei 3 min; Gradient: 0.0 min 30% B -> 5.5 min 30% B -> 17.65 min 95% B -> 19.48 min 95% B -> 19.66 min 30% B -> 20.51 min 30% B; Fluss: 75 ml/min. UV-Detektion: 210 nm
Methode 18 (präparative HPLC):
Säule: Chromatorex C18, 10 μηη, 125 x 30 mm; Eluent A: Wasser, Eluent B: Acetonitril; Injektion bei 3 min; Gradient: 0.0 min 10% B -> 5.5 min 10% B -> 17.65 min 95% B -> 19.48 min 95% B -> 19.66 min 10% B -> 20.51 min 10% B; Fluss: 75 ml/min. UV-Detektion: 210 nm
Methode 19 (präparative HPLC):
Säule: Chromatorex C18, 10 μιτι, 250 x 40 mm; Eluent A: Wasser, Eluent B: Acetonitril; Injektion bei 3 min; Gradient: 0.0 min 10% B -> 6.0 min 10% B -> 27 min 95% B -> 38 min 95% B -> 39 min 10% B -> 40.2 min 10% B; Fluss: 50 ml/min. UV-Detektion: 210 nm
Methode 20 (präparative HPLC):
Säule: Chromatorex C18, 10 μιτι, 250 x 40 mm; Eluent A: Wasser + 0.1 % TFA, Eluent B: Acetonitril; Injektion bei 3 min; Gradient: 0.0 min 10% B -> 6.0 min 10% B -> 27 min 95% B -> 38 min 95% B -> 39 min 10% B -> 40.2 min 10% B; Fluss: 50 ml/min. UV-Detektion: 210 nm
Methode 21 (präparative HPLC):
Säule: Reprosil C18, 10 μιτι, 250 x 40 mm; Eluent A: Wasser + 0.1 % TFA, Eluent B: Acetonitril; Injektion bei 3 min; Gradient: 0.0 min 10% B -> 6.0 min 10% B -> 27 min 95% B -> 38 min 95% B -> 39 min 10% B -> 40.2 min 10% B; Fluss: 50 ml/min. UV-Detektion: 210 nm.
Methode 22 (präparative HPLC):
Säule: Reprosil C18, 10 μιτι, 125 x 30 mm; Eluent A: Wasser + 0.1 % TFA, Eluent B: Acetonitril; Injektion bei 3 min; Gradient: 0.0 min 10% B -> 5.5 min 10% B -> 17.65 min 95% B -> 19.48 min 95% B -> 19.66 min 10% B -> 20.51 min 10% B; Fluss: 75 ml/min. UV-Detektion: 210 nm.
Methode 23 (präparative HPLC):
Chromatorex C18, 10 μιτι, 125 x 30 mm; Eluent A: Wasser + 0.1 % TFA, Eluent B: Acetonitril; Injektion bei 3 min; Gradient: 0.0 min 10% B -> 5.5 min 10% B -> 17.65 min 95% B -> 19.48 min 95% B -> 19.66 min 10% B -> 20.51 min 10% B; Fluss: 75 ml/min. UV-Detektion: 210 nm
Methode 24 (präparative HPLC):
Säule: Reprosil C18, 10 μιτι, 125 x 30 mm; Eluent A: Wasser + 0.1 % TFA, Eluent B: Acetonitril; Injektion bei 3 min; Gradient: 0.0 min 30% B -> 5.5 min 30% B -> 17.65 min 95% B -> 19.48 min 95% B -> 19.66 min 30% B -> 20.51 min 30% B; Fluss: 75 ml/min. UV-Detektion: 210 nm
Methode 25 (präparative HPLC):
Instrument: Waters Acquity SQD UPLC System; Säule: Waters Acquity UPLC HSS T3 1.8 μ, 50 x 1 mm; Eluent A: 1 I Wasser + 0.25 ml 99%-ige Ameisensäure, Eluent B: 1 I Acetonitril + 0.25 ml 99%-ige Ameisensäure; Gradient: 0.0 min 90% A -> 1.2 min 5% A -> 2.0 min 5% A; Ofen: 50°C; Fluss: 0.40 ml/min; UV-Detektion: 208-400 nm.
Methode 26 (präparative HPLC):
Instrument: Waters Prep LC/MS System, Säule: Phenomenex Kinetex C18, 5 μηη 100 x 30 mm; At-Column Injektion (Komplettinjektion); Eluent A: Wasser, Eluent B: Acetonitril, Eluent C: 2% Ameisensäure in Wasser; Fluss für Eluent (A + B): 65 ml/min, Fluss für Eluent C: konstant 5 ml/min; Gradient (A / B): 0.0 min 30% B -> 2 min 30% B -> 2.2 min 50% B -> 7 min 90% B -> 7.5 min 92% B -> 9 min 92% B -> 30% B; UV-Detektion: 210 nm.
Methode 27 (präparative HPLC):
Säule: Chromatorex, C18, 10 μηη, 125 mm x 30 mm; Eluent A: Wasser, Eluent B: Acetonitril; Injektion bei 3 min; Gradient: 0.0 min 10% B -> 6 min 10% B -> 27 min 95% B -> 38 min 95% B -^ 39 min 10% B ^ 40 min 10% B; Fluss: 75 ml/min, UV-Detektion: 210 nm.
Methode 28 (präparative HPLC):
Säule: Reprosil C18 10 μηη, 250 mm x 40 mm; Eluens A: Wasser + 0.1 % Ameisensäure, Eluens B: Acetonitril + 0.1 % Ameisensäure; Gradient: 0.0 min 10% B -> 6 min 10% B -> 27 min 95% B -> 38 min 95% B -> 39 min 10% B -> 40 min 10% B; Fluss: 75 ml/min, UV-Detektion: 210 nm. Methode 29 (präparative HPLC):
Säule: Chromatorex C18, 10 μηη, 250 mm x 40 mm; Eluent A: Wasser + 0.1 % Ameisensäure, Eluent B: Methanol + Ameisensäure; Gradient: 0.0 min 20% B -> 6.2 min 20% B -> 6.5 min 40% B -> 15.5 min 60% B -> 16 min 100% B -> 23 min 100% B -> 23.6 min 20% B -> 25.8 min 20% B; Fluss: 75 ml/min, UV-Detektion: 210 nm.
Methode 30 (präparative HPLC):
Säule: Chromatorex C18, 10 μηη, 125 mm x 30 mm; Eluent A: Wasser + 0.1 % Ameisensäure, Eluent B: Acetonitril + 0.1 % Ameisensäure; Injektion bei 3 min; Gradient: 0.0 min 10% B— > 6.0 min 10% B -> 27 min 95% B -> 38 min 95% B -> 39 min 10% B -> 40 min 10% B; Fluss: 75 ml/min, UV-Detektion: 210 nm.
Methode 31 (präparative HPLC):
Säule: Chromatorex C18 10 μηη, 250 mm x 40 mm; Eluens A: Wasser + 0.1 % Ameisensäure, Eluens B: Acetonitril + 0.1 % Ameisensäure; Gradient: 0.0 min 10% B -> 6.0 min 10% B -> 27
min 95% B -> 38 min 95% B -> 39 min 10% B -> 40 min 10% B; Fluss: 75 ml/min, UV-Detektion: 210 nm.
Methode 32 (präparative HPLC):
Säule: Reprosil C18 10 μηη, 250 mm x 40 mm; Eluens A: Wasser + 0.1 % Ameisensäure, Eluens B: Methanol + Ameisensäure; Gradient: 0.0 min 5% B -> 5.0 min 5% B -> 5.5 min 20% B -> 10.4 min 40% B -> 10.9 min 100% B -> 18.9 min 100% B -> 19.2 min 5% B -> 22.4 min 5% B; Fluss: 75 ml/min, UV-Detektion: 210 nm.
Weitere Angaben:
Die Prozentangaben in den folgenden Beispiel- und Testbeschreibungen sind, sofern nicht an- ders angegeben, Gewichtsprozente; Teile sind Gewichtsteile. Lösungsmittelverhältnisse, Verdünnungsverhältnisse und Konzentrationsangaben von flüssig/flüssig-Lösungen beziehen sich jeweils auf das Volumen.
Bei Aufreinigungen von erfindungsgemäßen Verbindungen per präparativer HPLC nach den beschriebenen Methoden, in denen die Elutionsmittel Zusatzstoffe wie beispielsweise Trifluoressig- säure, Ameisensäure oder Ammoniak enthalten, können die erfindungsgemäßen Verbindungen in Salz-Form, beispielsweise als Trifluoracetat, Formiat oder Ammonium-Salz anfallen, sofern die erfindungsgemäßen Verbindungen ausreichend basische bzw. säure Funktionalitäten enthalten. Ein solches Salz kann durch verschiedene dem Fachmann bekannte Methoden in die entsprechende freie Base bzw. Säure überführt werden.
Reinheitsangaben beziehen sich in der Regel auf entsprechende Peak-Integrationen im LC/MS- Chromatogramm, können aber zusätzlich auch unter Zuhilfenahme des 1H-NMR-Spektrums ermittelt worden sein. Verbindungen können noch Lösungsmittelreste enthalten, welche bei der Angabe der Reinheit nicht notwendigerweise berücksichtigt wurde. Wenn keine Reinheit angegeben ist, handelt es sich entweder um eine 100%-Reinheit laut automatischer Peak-Integration im LC/MS-Chromatogramm oder die Reinheit wurde nicht explizit ermittelt.
Angaben zu Ausbeuten in % d. Th. sind in der Regel reinheitskorrigiert, sofern eine Reinheit < 100% angegeben ist. Bei lösungsmittelhaltigen oder verunreinigten Chargen kann die Ausbeute formal ">100%" betragen; in diesen Fällen ist die Ausbeute nicht lösungsmittel- bzw. reinheitskorrigiert.
Die nachfolgenden Beschreibungen der Kopplungsmuster von 1H-NMR-Signalen wurden teilweise direkt den Vorschlägen des ACD SpecManagers (ACD/Labs Release 12.00, Product version 12.5) oder ACD/Spectrus Processor 2014 (File Version S20S41 , Build 72444, 21 Aug 2014) oder ACD/Spektrus Processor 2015 Pack 2 (File Version S40S41 , Build 79720, 30 Jul 2015) entnommen und nicht notwendigerweise streng hinterfragt. Teilweise wurden die Vorschläge des Spec- Managers manuell angepasst. Manuell angepasste bzw. zugewiesene Beschreibungen orientieren
sich in der Regel an dem optischen Erscheinungsbild der betreffenden Signale und entsprechen nicht notwendigerweise einer strengen, physikalisch korrekten Interpretation. In der Regel bezieht sich die Angabe zur chemischen Verschiebung auf das Zentrum des betreffenden Signals. Bei breiten Multipletts erfolgt die Angabe eines Intervalls. Durch Lösungsmittel oder Wasser verdeckte oder teilweise verdeckte Signale wurden entweder tentativ zugeordnet oder sind nicht aufgeführt. Stark verbreiterte Signale - z.B. verursacht durch schnelle Rotation von Molekülteilen oder aufgrund von austauschenden Protonen - wurden ebenfalls tentativ zugeordnet (oft als breites Mul- tiplett oder breites Singulett bezeichnet) oder sind nicht aufgeführt.
Die 1H-NMR-Daten ausgewählter Beispiele werden teilweise in Form von 1H-NMR-Peaklisten notiert. Zu jedem Signalpeak wird erst der δ-Wert in ppm und dann die Signalintensität in runden Klammem aufgeführt. Die δ-Wert-Signalintensitäts-Zahlenpaare von verschiedenen Signalpeaks werden durch Kommata voneinander getrennt aufgelistet. Die Peakliste eines Beispieles hat daher die Form: δι (Intensitäti), 62 (lntensität.2), ... , δ, (Intensität), ... , δη (Intensität).
Die Intensität scharfer Signale korreliert mit der Höhe der Signale in einem gedruckten Beispiel ei- nes NMR-Spektrums in cm und zeigt im Vergleich mit anderen Signalen die wirklichen Verhältnisse der Signalintensitäten. Bei breiten Signalen können mehrere Peaks oder die Mitte des Signals und ihre relative Intensität im Vergleich zum intensivsten Signal im Spektrum gezeigt werden. Die Listen der 1H-NMR-Peaks sind ähnlich den klassischen 1H-NMR-Ausdrucken und enthalten somit gewöhnlich alle Peaks, die bei einer klassischen NMR-Interpretation aufgeführt werden. Darüber hinaus können sie wie klassische 1H-NMR-Ausdrucke Lösungsmittelsignale, Signale von Stereoisomeren der Zielverbindungen, die ebenfalls Gegenstand der Erfindung sind, und/oder Peaks von Verunreinigungen zeigen. Die Peaks von Stereoisomeren der Targetverbindungen und/oder Peaks von Verunreinigungen haben gewöhnlich im Durchschnitt eine geringere Intensität als die Peaks der Zielverbindungen (zum Beispiel mit einer Reinheit von >90%). Solche Stereoisomere und/oder Verunreinigungen können typisch für das jeweilige Herstellungsverfahren sein. Ihre Peaks können somit dabei helfen, die Reproduktion unseres Herstellungsverfahrens anhand von "Nebenprodukt-Fingerabdrücken" zu erkennen. Ein Experte, der die Peaks der Zielverbindungen mit bekannten Verfahren (MestreC, ACD-Simulation, oder unter Verwendung von empirisch ausgewerteten Erwartungswerten) berechnet, kann je nach Bedarf die Peaks der Zielverbindungen isolieren, wobei gegebenenfalls zusätzliche Intensitätsfilter eingesetzt werden. Diese Isolierung wäre ähnlich dem betreffenden Peak-Picking bei der klassischen 1H-NMR-lnterpretation.
Eine detaillierte Beschreibung der Darstellung von NMR-Daten in Form von Peaklisten kann der Publikation "Citation of NMR Peaklist Data within Patent Applications" entnommen werden (vgl. Research Disclosure Database Number 605005, 2014, 1. August 2014 oder http://www.researchdisclosure.com/searching-disclosures). In der Peak Picking Routine, die in der Research Disclosure Database Number 605005 beschrieben ist, kann der Parameter "Mini- mumHeight" zwischen 1 % und 4% eingestellt werden. Abhängig von der Art der chemischen
Struktur und/oder abhängig von der Konzentration der zu vermessenden Verbindung kann es sinnvoll sein, den Parameter "MinimumHeight" auf werte <1 % einzustellen.
Schmelzpunkte und Schmelzbereiche, soweit angegeben, sind nicht korrigiert.
Für alle Reaktanden oder Reagenzien, deren Herstellung im Folgenden nicht explizit beschrie- ben ist, gilt, dass sie von allgemein zugänglichen Quellen kommerziell bezogen wurden. Für alle übrigen Reaktanden oder Reagenzien, deren Herstellung im Folgenden ebenfalls nicht beschrieben ist und die nicht kommerziell erhältlich waren oder von Quellen bezogen wurden, die nicht allgemein zugänglich sind, ist ein Verweis auf die veröffentlichte Literatur angegeben, in der ihre Herstellung beschrieben ist. Ausgangsverbindungen und Intermediate:
Beispiel 1A
6-Brom-3-methyl-2-phenylchinolin-4-carbonylchlorid
Eine Suspension aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (10.0 g, 29.2 mmol, dar- stellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in Dichlormethan (100 ml) wurde mit einigen Tropfen DMF versetzt. Anschließend wurde Oxalylchlorid (5.1 ml, 58 mmol) langsam hinzugetropft. Es wurde bis zum Abklingen der Gasentwicklung gerührt und das Gemisch dann eingeengt und im Vakuum getrocknet. Der erhaltene Rückstand wurde direkt (ohne weitere Aufarbeitung) für Folgechemie eingesetzt. Beispiel 2A
(6-Chlor-2,3-difluorphenyl)methanol
Eine Lösung aus 6-Chlor-2,3-difluorbenzaldehyd (5.00 g, 28.3 mmol) in THF (20 ml) wurde bei RT portionsweise mit Natriumborhydrid (1.39 g, 36.8 mmol) versetzt (Gasentwicklung). Anschließend wurden weitere 20 ml THF hinzugegeben, und das Gemisch wurde 45 min bei RT gerührt. Danach
wurde das Gemisch mit Dichlormethan (100 ml), Wasser (100 ml) und gesättigter Ammoniumchlorid-Lösung (50 ml) versetzt und geschüttelt. Die wässrige Phase wurde zwischendurch mit konzentrierter Essigsäure angesäuert. Nach Phasentrennung wurde die organische Phase einmal mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde kurz im Vakuum getrocknet. Es wurden 5.35 g (100% Reinheit,„>106% d. Th.", nicht ganz trocken) der Titelverbindung erhalten.
GC-MS (Methode 12): Rt = 3.23 min, MS (Elpos): m/z = 178 [M]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 7.45 (dd, 1 H), 7.36 (ddd, 1 H), 5.36 (t, 1 H), 4.59 (dd, 3H).
Beispiel 3A
[2-(Difluormethoxy)-6-fluorphenyl]methanol
Eine Lösung aus 2-(Difluormethoxy)-6-fluorbenzaldehyd (10.0 g, 52.6 mmol) in Ethanol (100 ml) wurde unter Eiskühlung portionsweise mit Natriumborhydrid (995 mg, 26.3 mmol) versetzt, wobei die Innentemperatur auf ca. 20 °C anstieg. Anschließend wurde das Gemisch weitere 2 h ohne Kühlung gerührt. Danach wurde das Gemisch mit Dichlormethan (100 ml), Wasser (150 ml) und gesättigter Ammoniumchlorid-Lösung (50 ml) versetzt und geschüttelt. Das Gemisch wurde mit konzentrierter Essigsäure (ca. 1 ml) angesäuert. Nach Phasentrennung wurde die wässrige Phase einmal mit Dichlormethan (100 ml) extrahiert, und die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde kurz im Vakuum getrocknet. Es wurden 9.90 g (99% Reinheit, 97% d. Th.) der Titelverbindung erhalten.
GC-MS (Methode 12): Rt = 3.04 min, MS (Elpos): m/z = 192 [M]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.53), 0.008 (0.55), 0.918 (0.70), 1.909 (1.97), 4.488 (14.37), 4.492 (15.14), 4.501 (15.42), 4.505 (15.39), 5.105 (7.08), 5.1 19 (14.1 1 ), 5.133 (6.48), 7.017 (8.05), 7.035 (6.81 ), 7.056 (7.70), 7.099 (4.02), 7.121 (8.26), 7.142 (4.75), 7.202 (16.00), 7.386 (1 1.15), 7.403 (4.54), 7.407 (7.33), 7.423 (7.21 ), 7.428 (3.89), 7.444 (3.19).
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 7.50-6.97 (m, 4H), 5.12 (t, 1 H), 4.50 (dd, 2H).
Beispiel 4A
2-(Brommethyl)-1-chlor-3,4-difluorbenzol
Zu einer Lösung aus (6-Chlor-2,3-difluorphenyl)methanol (5.34 g, 29.9 mmol, Beispiel 2A) in Dichlormethan (30 ml) wurde bei -15 °C Phosphortribromid (1.6 ml, 16 mmol) unter Rühren tropfenweise gegeben. Anschließend wurde das Kältebad entfernt, und das Gemisch wurde weitere 2 h gerührt. Danach wurde das Gemisch langsam mit gesättigter Natriumhydrogencarbonat- Lösung, Wasser und Dichlormethan (jeweils 50 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die organische Phase mit gesättigter wässriger Natriumchlorid-Lösung (100 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde kurz im Vakuum getrocknet. Es wurden 3.88 g (94% Reinheit laut GC-MS, 51 % d. Th.) der Titelver- bindung erhalten.
GC-MS (Methode 12): Rt = 3.57 min, MS (Elpos): m/z = 240 [M]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 7.55 (dd, 1 H), 7.45 (ddd, 1 H), 4.71 (d, 3H).
Beispiel 5A
(+/-)-2-(3-Chlorpyridin-2-yl)propannitril (Racemat)
Zu einer Lösung aus 2,3-Dichlorpyridin (4.00 g, 27.0 mmol) und Propionitril (2.68 g, 48.7 mmol) in THF (50 ml) unter Argon wurde tropfenweise bei RT eine 1 M Lösung von LiHMDS in THF/Ethylbenzol (100 ml, 100 mmol) gegeben, und das Gemisch wurde 20 h bei RT gerührt. Anschließend wurde das Gemisch mit Wasser versetzt und mit Ethylacetat extrahiert. Die orga- nische Phase wurde über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde zunächst mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Dichlormethan/Methanol 95:5, Isolera One) vorgereinigt und danach mittels präparativer HPLC (Methode 15) nachgereinigt. Es wurden 2.23 g (80% Reinheit, 40% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.29 min; MS (ESIpos): m/z = 167 [M+H]+
Aus einem analog durchgeführten Experiment wurde folgendes H-NMR der Titelverbindung erhalten:
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.59 (dd, 1 H), 8.03 (dd, 1 H), 7.48 (dd, 1 H), 4.73 (q, 1 H), 1.59 (d, 3H).
Beispiel 6A
(+/-)-2-(5-Fluor-2-methoxyphenyl)propannitril (Racemat)
Zu einer Lösung aus Diisopropylamin (3.3 ml, 23 mmol) in THF (1 10 ml) wurde bei -78 °C eine 1.6 M Butyllithium-Lösung in Hexan (15 ml, 23 mmol) tropfenweise hinzugegeben, und das Gemisch wurde 45 min bei -78 °C gerührt. Anschließend wurde eine Lösung aus (5-Fluor-2- methoxyphenyl)acetonitril (3.50 g, 21.2 mmol, CAS-RN 501008-41-9, kommerziell verfügbar) in THF (1 10 ml) tropfenweise bei -78 °C hinzugegeben, und das Gemisch wurde eine weitere Stunde bei -78 °C gerührt. Anschließend wurde eine Lösung aus lodmethan (1.4 ml, 22 mmol) in THF (15 ml) langsam hinzugegeben, und das Gemisch wurde weitere 10 min bei -78 °C gerührt. Anschließend wurde das Kältebad entfernt, und das Gemisch wurde 1.5 h bei RT gerührt. Das Gemisch wurde mit gesättigter Ammoniumchlorid-Lösung versetzt und geschüttelt. Nach Zugabe von Ethylacetat wurden die Phasen getrennt und die wässrige Phase zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid- Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde mittels Flash-Säulenchromatographie (120 g Kieselgel Reveleris, Fluss 80 ml/min, Hep- tan/Ethylacetat-Gradient 1 :0— > 6:4, Laufzeit 38 min) gereinigt. Es wurden 2.40 g (97% Reinheit, 61 % d. Th.) der Titelverbindung erhalten.
GC-MS (Methode 13): Rt = 2.99 min; MS (ESIpos): m/z = 179 [M]+
1H-NMR (400 MHz, CDCI3): δ [ppm] = 7.20-7.10 (m, 1 H), 7.05-6.96 (m, 1 H), 6.85-6.81 (m, 1 H), 2.22 (q, 1 H), 3.84 (s, 3H), 1.57 (d, 3H).
Beispiel 7A
(+/-)-2-(3,5-Difluor-2-methoxyphenyl)propannitril (Racemat)

Zu einer Lösung aus Diisopropylamin (1.9 ml, 14 mmol) in THF (25 ml) wurde bei -78 °C eine 1.6 M Butyllithium-Lösung in Hexan (8.2 ml, 13 mmol) tropfenweise hinzugegeben, und das Gemisch wurde 30 min bei -78 °C gerührt. Anschließend wurde eine Lösung aus (3,5-Difluor-2- methoxyphenyl)acetonitril (2.00 g, 10.9 mmol, CAS-RN 886761-64-4, kommerziell verfügbar) in THF (5.5 ml) tropfenweise bei -78 °C hinzugegeben, und das Gemisch wurde weitere 30 min bei -78 °C gerührt. Anschließend wurde eine Lösung aus lodmethan (710 μΙ, 1 1 mmol) in THF (25 ml) langsam hinzugegeben, und das Gemisch wurde weitere 30 min bei -78 °C gerührt. Anschließend wurde das Gemisch mit gesättigter Ammoniumchlorid-Lösung versetzt und geschüttelt. Das Kältebad wurde entfernt, und das Gemisch wurde auf RT kommen gelassen. Durch Zu- gäbe von 1 M Salzsäure wurde die wässrige Phase auf ca. pH 3 eingestellt. Nach Zugabe von gesättigter Natriumchlorid-Lösung wurde das Gemisch dreimal mit Ethylacetat extrahiert, und die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels Flash-Säulenchromatographie (40 g Kieselgel, Laufmittel Dichlormethan) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vaku- um getrocknet. Es wurden 1.99 g (88% Reinheit, 81 % d. Th.) der Titelverbindung erhalten.
GC-MS (Methode 13): Rt = 2.97 min; MS (ESIpos): m/z = 179 [M]+
Beispiel 8A
(+/-)-2-(4-Fluor-2-methoxyphenyl)propannitril (Racemat)

Zu einer Lösung aus 1-(4-Fluor-2-methoxyphenyl)ethanon (5.00 g, 29.7 mmol, CAS-RN 51788-80- 8, kommerziell verfügbar) in 1 ,2-Dimethoxyethan (30 ml) unter Stickstoff wurde TosMIC (6.39 g, 32.7 mmol) gegeben, und das Gemisch wurde auf -10 °C abgekühlt. Anschließend wurde eine Lösung aus Kalium-ierf-butylat (6.67 g, 59.5 mmol) in ierf-Butanol (75 ml) langsam hinzugegeben, wobei die Temperatur unter 5 °C gehalten wurde. Nach 2 h Rühren bei 0 °C und 1 h bei RT wurde die erhaltene Suspension bis zum Erhalt eines Schlammes aufkonzentriert und mit Wasser (60 ml) versetzt. Nach dreimaligem Extrahieren mit Diethylether (jeweils 50 ml) wurden die vereinigten organischen Phasen über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels Flash-Säulenchromatographie (120 g Kieselgel, Laufmittel Dichlormethan, Fluss 40 ml/min) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrock- net. Es wurden 3.77 g (99% Reinheit, 70% d. Th.) der Titelverbindung erhalten.
GC-MS (Methode 13): Rt = 2.97 min; MS (ESIpos): m/z = 179 [M]+
Beispiel 9A
(+/-)-2-(2-Fluor-6-methoxyphenyl)propannitril (Racemat)
Zu einer Lösung aus Λ/,/V-Diisopropylethylamin (5.8 ml, 33 mmol) in THF (75 ml) unter Argon wurde bei -78 °C ein Gemisch aus 2.5 M Butyllithium-Lösung in Hexan (13 ml, 33 mmol) und THF (50 ml) langsam hinzugegeben. Das Reaktionsgemisch wurde auf 0 °C kommen gelassen, 5 min bei 0 °C gerührt und wieder auf -78 °C abgekühlt. Dann wurde eine Lösung aus (2-Fluor- 6-methoxyphenyl)acetonitril (5.00 g, 30.3 mmol, CAS-RN 500912-18-5, kommerziell verfügbar) in THF (25 ml) langsam hinzugegeben. Das Reaktionsgemisch wurde erneut auf 0 °C kommen gelassen, 5 min bei 0 °C gerührt und wieder auf -78 °C abgekühlt. Sodann wurde eine Lösung aus lodmethan (2.0 ml, 31.79 mmol) in THF (25 ml) langsam hinzugegeben. Das Gemisch wurde über Nacht gerührt, wobei die Temperatur langsam auf RT kommen gelassen wurde. Anschließend wurde das Gemisch bei 0 °C langsam mit gesättigter Ammoniumchlorid-Lösung und Wasser (jeweils 50 ml) versetzt, geschüttelt und zweimal mit Ethylacetat (jeweils 150 ml) extra- hiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung (200 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (400 g Kieselgel Büchi Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat 9:1 ) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand (kurz) im Vakuum getrocknet. Es wurden 3.33 g (100% Reinheit, 61 % d. Th.) der Titelverbindung erhalten.
GC-MS (Methode 12): Rt = 4.31 min; MS (ESIpos): m/z = 179 [M]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.492 (15.85), 1.509 (16.00), 3.314 (9.13), 3.887 (0.90), 4.452 (0.69), 4.455 (0.70), 4.470 (2.12), 4.473 (2.10), 4.488 (2.1 1 ), 4.491 (2.05), 4.505 (0.69), 4.508 (0.65), 6.861 (1.62), 6.863 (1.60), 6.884 (2.45), 6.907 (1.81 ), 6.909 (1.81 ), 6.943 (3.36), 6.964 (3.72), 7.353 (1.56), 7.370 (1.87), 7.374 (2.99), 7.391 (3.02), 7.395 (1.60), 7.412 (1.36).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 7.44-7.30 (m, 1 H), 6.95 (d, 1 H), 6.92-6.85 (m, 1 H), 4.48 (qd, 1 H), 3.88 (s, 3H), 1.50 (d, 3H).
Beispiel 10A
(+/-)-2-(2-Chlor-5-fluorphenyl)propannitril (Racemat)
Zu einer Lösung aus Λ/,/V-Diisopropylethylamin (2.8 ml, 16 mmol) in THF (40 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2.5 M Butyllithium-Lösung in Hexan (6.5 ml, 16 mmol), verdünnt mit THF (20 ml), zugetropft. Die Gemisch wurde danach unter Rühren auf 0 °C kommen gelassen und nach 5 min wieder auf -78 °C gekühlt. Anschließend wurde eine Lösung aus (2-Chlor-5-fluorphenyl)acetonitril (2.50 g, 14.7 mmol, CAS-RN 395675-23-7, kommerziell verfügbar) in THF (20 ml) langsam hinzugetropft. Die Gemisch wurde danach unter Rühren auf 0 °C kommen gelassen und nach 5 min wieder auf -78 °C gekühlt. Anschließend wurde eine Lösung aus lodmethan (960 μΙ, 15 mmol) in THF (20 ml) langsam hinzugetropft. Das Gemisch wurde über Nacht weiter gerührt, wobei das Kältebad (Trockeneis/Isopropanol) langsam auf RT kommen gelassen wurde. Anschließend wurde das Gemisch bei 0 °C langsam mit gesättigter Ammoniumchlorid-Lösung und Wasser (jeweils 50 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase zweimal mit Ethylacetat (jeweils 150 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung (200 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat-Gradient 93:7— > 7:3, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 2.54 g (97% Reinheit, 91 % d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.78 min; MS (ESIpos): m/z = 184 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 7.60 (dd, 1 H), 7.48 (dd, 1 H), 7.31 (td, 1 H), 4.52 (q, 1 H), 1.59 (d, 3H).
Beispiel 11 A
(2-Chlor-3,6-difluorphenyl)acetonitril
Zu einer Lösung aus 2-(Brommethyl)-3-chlor-1 ,4-difluorbenzol (4.80 g, 19.9 mmol, CAS-RN 90292-67-4, kommerziell verfügbar) in Dichlormethan (40 ml) wurden unter Rühren Wasser (40
ml) und Tetrabutylammoniumbromid (641 mg, 1.99 mmol) gegeben. Anschließend wurde eine Lösung aus Kaliumcyanid (3.88 g, 59.6 mmol) in Wasser (40 ml) hinzugegeben, und das Gemisch wurde 2.5 h bei RT gerührt. Anschließend wurden die Phasen getrennt, und die organische Phase wurde dreimal mit gesättigter Natriumhydrogencarbonat-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde kurz im Vakuum getrocknet. Es wurden 3.80 g (94% Reinheit, 96% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.59 min; MS (ESIneg): m/z = 186 [M-H]"
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 7.57 (td, 1 H), 7.44 (td, 1 H), 4.15 (d, 2H).
Beispiel 12A
(2-Chlor-3-fluorphenyl)acetonitril
Zu einer Lösung aus 1 -(Brommethyl)-2-chlor-3-fluorbenzol (6.96 g, 31.1 mmol) in Dichlormethan (60 ml) wurden unter Rühren Wasser (60 ml) und Tetrabutylammoniumbromid (1.00 g, 3.1 1 mmol) gegeben. Anschließend wurde eine Lösung aus Kaliumcyanid (6.08 g, 93.4 mmol) in Wasser (60 ml) hinzugegeben, und das Gemisch wurde 2.5 h bei RT gerührt. Anschließend wurden die Phasen getrennt, und die organische Phase wurde dreimal mit gesättigter Natriumhydrogencarbonat-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde kurz im Vakuum getrocknet. Es wurden 4.98 g (100% Reinheit, 94% d. Th.) der Titelverbindung erhalten.
GC-MS (Methode 12): Rt = 4.03 min, MS (Elpos): m/z = 169 [M]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 4.175 (16.00), 7.400 (0.90), 7.408 (1.10), 7.414 (1.15), 7.417 (1.13), 7.424 (2.60), 7.440 (2.84), 7.443 (2.99), 7.459 (5.37), 7.468 (1.52), 7.474 (1.39).
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 7.50-7.37 (m, 3H), 4.18 (s, 2H).
Beispiel 13A
(6-Chlor-2,3-difluorphenyl)acetonitril
Zu einer Lösung aus 2-(Brommethyl)-1-chlor-3,4-difluorbenzol (3.87 g, 16.0 mmol, Beispiel 4A) in Acetonitril (48 ml) wurden unter Rühren Trimethylsilylcyanid (2.5 ml, 18 mmol) und eine 1 M Lösung aus Tetrabutylammoniumfluorid in THF (19 ml, 19 mmol) gegeben, und das Gemisch wurde 30 min bei 80 °C gerührt. Nach Abkühlen auf RT wurde das Lösungsmittel am Rotations- Verdampfer entfernt. Der Rückstand wurde in Ethylacetat (80 ml) aufgenommen, und die Lösung wurde mit Wasser und gesättigter Natriumchlorid-Lösung (jeweils 80 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat-Gradient 93:7— > 7:3, Isolera One) gereinigt. Die vereinigten Zielfraktio- nen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 2.24 g (71 % Reinheit laut GC-MS, 96% d. Th.) der Titelverbindung erhalten.
GC-MS (Methode 12): Rt = 3.92 min, MS (Elpos): m/z = 187 [M]+
LC-MS (Methode 1 ): Rt = 1.57 min; MS (ESIneg): m/z = 186 [M-H]-
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 7.58 (dd, 1 H), 7.49 (ddd, 1 H), 4.16 (d, 2H). Beispiel 14A
(2,3,5,6-Tetrafluorphenyl)acetonitril
Kaliumcyanid (1.34 g, 20.6 mmol) wurde in Acetonitril (64 ml) und Wasser (13 ml) bei RT vorgelegt. 3-(Brommethyl)-1 ,2,4,5-tetrafluorbenzol (5.00 g, 20.6 mmol) wurde hinzugefügt und das Ge- misch anschließend 5 h bei 40 °C gerührt, dann über Nacht bei RT weitergerührt. Zur Aufarbeitung wurde die Reaktionsmischung mit 150 ml Wasser verdünnt, mit ca. 10 ml 1 M Natronlauge auf pH 13 gestellt und zweimal mit ierf-Butylmethylether extrahiert. Die vereinigten organischen Phasen wurden zweimal mit einer gesättigten Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet und am Rotationsverdampfer bei 80 mbar Vakuum und maximal 40 °C eingeengt. Der Rückstand wurde in wenig Dichlormethan gelöst und mittels Flash-Chromatographie (Isolera, 100 g Ultra Snap Säule, Cyclohexan / Ethylacetat Gradient) gereinigt. Die vereinigten Zielfraktionen wurden am Rotationsverdampfer bei 80 mbar Vakuum und maximal 40 °C eingeengt. Es wurden 2.47 g (100% Reinheit, 63% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 0.77 min; kaum Ionisierung
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.92), 0.008 (1.75), 2.524 (1.31 ), 4.221 (16.00), 7.924 (0.58), 7.943 (1.16), 7.951 (1.18), 7.963 (0.79), 7.970 (2.15), 7.977 (0.73), 7.989 (1.16), 7.997 (1.11 ), 8.016 (0.53).
Beispiel 15A
(+/-)-Ethyl-(2-chlorphenyl)(cyan)acetat (Racemat)
Zu einer Lösung aus (2-Chlorphenyl)acetonitril (75.0 g, 495 mmol) in Toluol (2.0 I) unter Argon wurden Natriumhydrid (21.8 g, 544 mmol, 60% in Mineralöl) und Diethylcarbonat (60 ml, 490 mmol) gegeben, und das Gemisch wurde über Nacht bei 80 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit 1 Liter 1 M Salzsäure versetzt, 5 min gerührt und dreimal mit Ethyl- acetat extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde mittels Flash-Säulenchromatographie (Kieselgel, Cyclohexan/Ethylacetat 10:1 ) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 100 g (90% d. Th.) der Titelverbindung erhalten. Eine alternative Darstellungsmethode ist beschrieben in Journal of the American Chemical Society 1965, 87 (5), 1 1 15-1 120.
Beispiel 16A
(+/-)-ie/f-Butyl-2-(3-chlorpyridin-2-yl)-2-cyanpropanoat (Racemat)

Zu einer Lösung aus (+/-)-2-(3-Chlorpyridin-2-yl)propannitril (1.00 g, 6.00 mmol, Beispiel 5A) in THF (8.8 ml) unter Argon wurde bei -78 °C langsam eine 2 M Lösung aus LDA in THF (4.5 ml, 9.0 mmol) hinzugegeben, und das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde Di-ie f-butyldicarbonat (1.96 ml, 9.00 mmol) langsam bei -78 °C hinzu getropft, das Kältebad wurde entfernt, und das Reaktionsgemisch wur- de bei RT über Nacht gerührt. Anschließend wurde das Gemisch langsam unter Rühren mit Wasser versetzt, geschüttelt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen
Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde mittels präparativer HPLC (Methode 22) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 921 mg (86% Reinheit, 49% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.95 min Beispiel 17A
(+/-)-ie/f-Butyl-2-cyan-2-(2,6-difluorphenyl)propanoat (Racemat)
Zu einer Lösung aus (+/-)-2-(2,6-Difluorphenyl)propannitril (5.00 g, 29.91 mmol, darstellbar gemäß Russian Journal of Organic Chemistry 2009, 45, 1531-1534) in THF (38 ml) unter Argon wurde bei -78 °C langsam eine 2 M Lösung aus LDA in THF (24 ml, 49 mmol) hinzugegeben, und das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde Di-ie/f-butyldicarbonat (10.7 g, 49.0 mmol) langsam bei -78 °C hinzugetropft, und das Gemisch wurde danach 3 h bei 0 °C gerührt. Anschließend wurde das Gemisch langsam unter Rühren mit Wasser versetzt, geschüttelt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 6.80 g (79% Reinheit, 66% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.08 min; MS (ESIpos): m/z = 268 [M+H]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.1 16 (2.15), 1.132 (2.17), 1.385 (1.30), 1.403 (13.17), 1.437 (16.00), 1.468 (5.97), 1.945 (1.50), 1.949 (2.52), 1.953 (1.48), 7.223 (0.50), 7.245 (0.67), 7.248 (0.58), 7.269 (0.62), 7.563 (0.41 ).
Beispiel 18A
(+/-)-ie/f-Butyl-cyan(3-methoxyphenyl)acetat (Racemat)

Zu einer Lösung aus (3-Methoxyphenyl)acetonitril (3.00 g, 20.4 mmol) in THF (16 ml) unter Argon wurde bei -78 °C langsam eine 2 M Lösung aus LDA in THF (15 ml, 31 mmol) hinzugegeben, und das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde Di-ie/f-butyldicarbonat (6.67 g, 30.6 mmol), gelöst in THF (15 ml), langsam bei -78 °C hinzugetropft, und das Gemisch wurde auf RT kommen gelassen und über Nacht bei RT gerührt. Anschließend wurde das Gemisch langsam unter Rühren mit Wasser versetzt, geschüttelt und zweimal mit Ethylacetat (60 ml und 45 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde im Vakuum getrock- net. Der Rückstand wurde mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap- Cartridge KP-Sil, Cyclohexan/Ethylacetat 100:0— > 9:1 ) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 4.00 g (78% Reinheit, 62% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.01 min; MS (ESIpos): m/z = 248 [M+H]+
Beispiel 19A
(+/-)-ie/f-Butyl-(3-chlorphenyl)(cyan)acetat (Racen
Zu einer Lösung aus (3-Chlorphenyl)acetonitril (3.00 g, 19.8 mmol) in THF (16 ml) unter Argon wurde bei -78 °C langsam eine 2 M Lösung aus LDA in THF (15 ml, 30 mmol) hinzugegeben, und das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde Di-ie f-butyldicarbonat (6.48 g, 29.7 mmol), gelöst in THF (15 ml), langsam bei -78 °C hinzugetropft, und das Gemisch wurde auf RT kommen gelassen und über Nacht bei RT gerührt. Anschließend wurde das Gemisch langsam unter Rühren mit Wasser versetzt, geschüttelt und zweimal mit Ethylacetat (60 ml und 40 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde im Vakuum getrocknet. Der Rückstand wurde mittels Flash-Säulenchromatographie (120 g Kieselgel Biotage Snap- Cartridge KP-Sil, Cyclohexan/Ethylacetat 100:0— > 9:1 ) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 400 mg (55% Reinheit, 4% d. Th.) einer ersten Charge der Titelverbindung, 2.6 g (80% Reinheit, 42% d. Th., siehe Analytik) einer zweiten Charge der Titelverbindung und 2.5 g (70% Reinheit, 35% d. Th.) einer drit-
ten Charge der Titelverbindung erhalten. Die drei Chargen wurden vereinigt und in der Folgereaktion (siehe Beispiel 86A) eingesetzt.
LC-MS (Methode 1 ): Rt = 2.07 min; MS (ESIneg): m/z = 250 [M-H]" Beispiel 20A
(+/-)-ie/f-Butyl-cyan(3-methylphenyl)acetat (Racemat)
Zu einer Lösung aus (3-Methylphenyl)acetonitril (3.00 g, 22.9 mmol) in THF (35 ml) unter Argon wurde bei -78 °C langsam eine 2 M Lösung aus LDA in THF (17 ml, 34 mmol) hinzugegeben, und das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abge- kühlt. Anschließend wurde Di-ie/f-butyldicarbonat (7.49 g, 34.3 mmol) langsam bei -78 °C hinzugetropft, und das Gemisch wurde 3 h bei 0 °C gerührt. Anschließend wurde das Gemisch langsam unter Rühren mit Wasser (50 ml) versetzt, geschüttelt und zweimal mit Ethylacetat (70 ml und 50 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde im Vakuum getrocknet. Der Rückstand wurde mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat 100:0— > 9:1 ) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 3.20 g (82% Reinheit, 50% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.07 min; MS (ESIneg): m/z = 230 [M-H]- Beispiel 21A
(+/-)-ie/f-Butyl-cyan(2-fluor-6-methoxyphenyl)acetat (Racemat)
Zu einer Lösung aus (2-Fluor-6-methoxyphenyl)acetonitril (5.00 g, 30.3 mmol, CAS-RN 500912- 18-5) in THF (25 ml) unter Argon wurde bei -65 °C langsam eine 2 M Lösung aus LDA in THF (23 ml, 45 mmol) hinzugegeben, und das Gemisch wurde auf 0 °C kommen gelassen und nach
15 min wieder auf -65 °C abgekühlt. Anschließend wurde eine Lösung aus Di-ie/f-butyldicarbonat (10 ml, 45 mmol) in THF (11 ml) langsam hinzugetropft, wobei die Innen-temperatur unter -40 °C gehalten wurde. Das Gemisch wurde über Nacht gerührt, wobei die Temperatur langsam auf RT kommen gelassen wurde. Anschließend wurde das Gemisch unter Eiskühlung langsam unter Rühren mit 1 M Salzsäure (45.4 ml, 45.4 mmol), gefolgt von Wasser (30 ml) und Ethylacetat (80 ml), versetzt. Nach Schütteln und Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (80 ml) extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (500 g Kieselgel, Cyclohexan/Ethylacetat 10:1 ) gereinigt. Die verei- nigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 10.4 g (62% Reinheit, 80% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.89 min; MS (ESIpos): m/z = 266 [M+H]+ Beispiel 22A
(+/-)-ie/f-Butyl-2-cyan-2-(2-methoxyphenyl)propanoat (Racemat)
Zu einer Lösung aus 2-(2-Methoxyphenyl)propannitril (2.00 g, 12.4 mmol, CAS-RN 621 15-71-3, kommerziell verfügbar) in THF (18 ml) unter Argon wurde bei -78 °C langsam eine 2 M Lösung aus LDA in THF (9.3 ml, 19 mmol) hinzugegeben, und das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde Di-ie/f- butyldicarbonat (4.06 g, 18.6 mmol) langsam hinzugegeben. Das Gemisch wurde über Nacht gerührt, wobei die Temperatur langsam auf RT kommen gelassen wurde. Anschließend wurde das Gemisch langsam unter Rühren mit Wasser versetzt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dich- lormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel, Cyclohexan/Ethylacetat 85:15) vorgereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde in Aceton itril/Wasser lyophilisiert. Anschließend wurde das vorgereinigte Produkt mittels präparativer HPLC (Säule: XBridge C18, 5 μηη, 100 mm x 30 mm; Fluss: 75 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 0.30 ml; Eluent: 40% Acetonitril / 55% Wasser / 5% (Wasser + 1 % Ammoniak) -> 70% Acetonitril / 25% Wasser / 5% (Wasser + 1 % Ammoniak; Laufzeit 5 min) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rück-
stand wurde in Aceton itril/Wasser lyophilisiert. Es wurden 1.44 g (100% Reinheit, 44% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.97 min; MS (ESIpos): m/z = 262 [M+H]+ Beispiel 23A
(+/-)-ie/f-Butyl-2-(2-chlor-6-fluorphenyl)-2-cyanpropanoat (Racemat)
Zu einer Lösung aus 2-(2-Chlor-6-fluorphenyl)propannitril (5.00 g, 27.2 mmol, CAS-RN 1260829- 70-6, kommerziell verfügbar) in THF (22 ml) unter Argon wurde bei -78 °C langsam eine 2 M Lösung aus LDA in THF (20 ml, 41 mmol) hinzugegeben, und das Gemisch wurde auf 0 °C kom- men gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde Di-ie/f- butyldicarbonat (9.4 ml, 41 mmol) langsam hinzugegeben. Das Gemisch wurde über Nacht gerührt, wobei die Temperatur langsam auf RT kommen gelassen wurde. Anschließend wurde das Gemisch langsam unter Rühren mit Wasser versetzt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewa- sehen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde zweimal mittels präparativer HPLC (Methode 14) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde in Aceton itril/Wasser lyophilisiert. Es wurden 1.33 g (98% Reinheit, 17% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 3): Rt = 3.49 min; MS (ESIpos): m/z = 284 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 7.59-7.43 (m, 2H), 7.37 (ddd, 1 H), 2.04 (d, 3H), 1.44 (s, 9H).
Beispiel 24A
(+/-)-ie/f-Butyl-3-(2-chlorphenyl)-3-cyanpropanoat (Racemat)

Zu einer Lösung aus (2-Chlorphenyl)acetonitril (5.00 g, 33.0 mmol) in THF (50 ml) unter Agon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (25 ml, 49 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde ie/f-Butyl-bromacetat (5.8 ml, 40 mmol) langsam unter Rühren hin- zu getropft. Das Kältebad wurde entfernt und das Gemisch unter Rühren über Nacht auf RT kommen gelassen. Anschließend wurde das Gemisch langsam mit Wasser versetzt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde mittels präparativer HPLC [Säule: Kinetix C18, 5 μηη, 100 x 21.2 mm; Fluss: 30 ml/ min; Detektion: 210 nm; Injektionsvolumen: 0.4 ml; Eluent: 50% Wasser / 45% Ace- tonitril / 5% Ameisensäure (1 % in Wasser)— > 5% Wasser / 90% Acetonitril / 5% Ameisensäure (1 % in Wasser), Laufzeit: 6.0 min] gereinigt. Es wurden 6.98 g (Reinheit 100%, 78% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.10 min; MS (ESIpos): m/z = 266 [M+H]+ Beispiel 25A
(+/-)-ie/f-Butyl-3-(3-chlorphenyl)-3-cyanpropanoat (Racemat)
Zu einer Lösung aus (3-Chlorphenyl)acetonitril (1 .00 g, 6.60 mmol) in THF (10 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (4.9 ml, 9.9 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde ie f-Butyl-bromacetat (1.5 ml, 9.9 mmol) langsam unter Rühren hinzu getropft. Das Kältebad wurde entfernt und das Gemisch unter Rühren über Nacht auf RT kommen gelassen. Anschließend wurde das Gemisch langsam mit Wasser versetzt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Das Rohprodukt wurde mit dem Rohprodukt aus einem analog erhaltenen Vorversuch (eingesetzte Menge an (3-Chlorphenyl)acetonitril: 100 mg (0.66 mmol)) vereinigt, in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap- Cartridge KP-Sil, Cyclohexan/Ethylacetat 97:3 und 8:2, Isolera One) gereinigt. Es wurden 710 mg (Reinheit 95%, 35% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.10 min; MS (ESIpos): m/z = 266 [M+H]+
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: 1.215 (0.55), 1.365 (16.00), 1.399 (0.56), 1.418 (1.40), 1.430 (0.66), 2.927 (0.56), 2.940 (0.56), 3.024 (0.54), 3.040 (0.55), 4.538 (0.40), 7.430 (0.56), 7.434 (0.78), 7.440 (1.60), 7.451 (0.49), 7.556 (0.75).
Beispiel 26A
(+/-)-fe/f-Butyl-3-cyan-3-(2-methylphenyl)propanoat (Racemat)
Zu einer Lösung aus (2-Methylphenyl)acetonitril (1.00 g, 7.62 mmol) in THF (1 1 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (5.7 ml, 1 1 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C ab- gekühlt. Anschließend wurde ie/f-Butyl-bromacetat (1.7 ml, 1 1 mmol) langsam unter Rühren hinzu getropft. Das Kältebad wurde entfernt und das Gemisch unter Rühren über Nacht auf RT kommen gelassen. Anschließend wurde das Gemisch langsam mit Wasser versetzt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat 97:3 -> 8:2, Isolera O- ne) gereinigt. Es wurden 880 mg (Reinheit 98%, 47% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.10 min; MS (ESIpos): m/z = 246 [M+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 7.47-7.39 (m, 1 H), 7.29-7.20 (m, 3H), 4.50 (dd, 1 H), 3.01-2.81 (m, 2H), 2.36 (s, 3H), 1.37 (s, 9H).
Beispiel 27A
(+/-)-ie/f-Butyl-3-cyan-3-(2,6-dichlorphenyl)propanoat (Racemat)
Zu einer Lösung aus (2,6-Dichlorphenyl)acetonitril (1.00 g, 5.38 mmol) in THF (8 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (4.0 ml, 8.1 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C
abgekühlt. Anschließend wurde ie/f-Butyl-bromacetat (1.2 ml, 8.1 mmol) langsam unter Rühren hinzu getropft. Das Kältebad wurde entfernt und das Gemisch unter Rühren über Nacht auf RT kommen gelassen. Anschließend wurde das Gemisch langsam mit Wasser versetzt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat 97:3 -> 8:2, Isolera O- ne) gereinigt. Es wurden 682 mg (98% Reinheit, 41 % d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.17 min; MS (ESIpos): m/z = 300 [M+H]+
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: 0.933 (1.68), 0.946 (1.79), 1.363 (16.00), 1.392 (2.64), 1.401 (1.37), 3.025 (0.58), 3.038 (0.57), 3.181 (0.57), 3.198 (0.58), 3.315 (0.99), 7.461 (0.61 ), 7.463 (0.68), 7.478 (0.76), 7.582 (2.03), 7.598 (1.35).
Beispiel 28A
(+/-)-ie/f-Butyl-3-cyan-3-(3-methoxyphenyl)propanoat (Racemat)
Zu einer Lösung aus (3-Methoxyphenyl)acetonitril (1.00 g, 6.79 mmol) in THF (10 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (5.1 ml, 10 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde ie f-Butyl-bromacetat (1.5 ml, 10 mmol) langsam unter Rühren hinzu getropft. Das Kältebad wurde entfernt und das Gemisch unter Rühren über Nacht auf RT kommen gelassen. Anschließend wurde das Gemisch langsam mit Wasser versetzt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat 97:3 -> 8:2, Isolera O- ne) gereinigt. Es wurden 389 mg (90% Reinheit, 20% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.96 min; MS (ESIpos): m/z = 262 [M+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 7.36-7.27 (m, 1 H), 7.05-6.98 (m, 2H), 6.94-6.87 (m, 1 H), 4.43 (dd, 1 H), 3.76 (s, 3H), 3.00 (dd, 1 H), 2.87 (dd, 1 H), 1.38 (s, 9H).
Beispiel 29A
(+/-)-fe/f-Butyl-3-(3-chlorpyridin-2-yl)-3-cyanbutanoat (Racemat)
Zu einer Lösung aus (+/-)-2-(3-Chlorpyridin-2-yl)propannitril (1.20 g, 7.20 mmol, Beispiel 5A) in THF (10 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (5.4 ml, 1 1 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde ie/f-Butyl-bromacetat (1.3 ml, 8.6 mmol) langsam unter Rühren hinzu getropft. Das Kältebad wurde entfernt und das Gemisch unter Rühren über Nacht auf RT kommen gelassen. Anschließend wurde das Gemisch langsam mit Was- ser versetzt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 22) gereinigt. Es wurden 1.08 g (100% Reinheit, 53% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.93 min; MS (ESIpos): m/z = 225 [M-'Bu+H]+ Beispiel 30A
(+/-)-ie/f-Butyl-3-(2-chlor-6-fluorphenyl)-3-cyanpropanoat (Racemat)
Zu einer Lösung aus (2-Chlor-6-fluorphenyl)acetonitril (1.00 g, 5.90 mmol) in THF (8.7 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (4.4 ml, 8.8 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde ief-Butyl-bromacetat (1.3 ml, 8.8 mmol) langsam unter Rühren hinzu getropft. Das Kältebad wurde entfernt und das Gemisch unter Rühren über Nacht auf RT kommen gelassen. Anschließend wurde das Gemisch langsam mit Wasser versetzt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat 97:3— > 8:2, Isolera One) ge-
reinigt. Es wurden 878 mg (98% Reinheit, 51% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.07 min; MS (ESIpos): m/z = 284 [M+H]+
1H-NMR (500 MHz, DMSO-d6): δ [ppm] = 7.54-7.47 (m, 1 H), 7.47-7.42 (m, 1 H), 7.37 (ddd, 1 H), 4.88-4.82 (m, 1 H), 3.14-3.07 (m, 1 H), 3.02-2.93 (m, 1 H), 1.34 (s, 9H). Beispiel 31A
(+/-)-ie/f-Butyl-3-cyan-3-(4-methylphenyl)propanoat (Racemat)
Zu einer Lösung aus (4-Methylphenyl)acetonitril (1.00 g, 7.62 mmol) in THF (1 1 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (5.7 ml, 1 1 mmol) ge- geben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde ie/f-Butyl-bromacetat (1.7 ml, 1 1 mmol) langsam unter Rühren hinzu getropft. Das Kältebad wurde entfernt und das Gemisch unter Rühren über Nacht auf RT kommen gelassen. Anschließend wurde das Gemisch langsam mit Wasser versetzt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat 97:3 -> 8:2, Isolera O- ne) gereinigt. Es wurden 288 mg (90% Reinheit, 14% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.09 min; MS (ESIpos): m/z = 246 [M+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 7.32 (d, 2H), 7.21 (d, 2H), 4.40 (dd, 1 H), 2.95 (dd, 1 H), 2.83 (dd, 1 H), 2.29 (s, 3H), 1.37 (s, 9H).
Beispiel 32A
(+/-)-ie/f-Butyl-3-cyan-3-(3-methylphenyl)propanoat (Racemat)
H

Zu einer Lösung aus (3-Methylphenyl)acetonitril (1.00 g, 7.62 mmol) in THF (1 1 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (5.7 ml, 1 1 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde ie/f-Butyl-bromacetat (1.7 ml, 1 1 mmol) langsam unter Rühren hin- zu getropft. Das Kältebad wurde entfernt und das Gemisch unter Rühren über Nacht auf RT kommen gelassen. Anschließend wurde das Gemisch langsam mit Wasser versetzt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat 97:3 -> 8:2, Isolera O- ne) gereinigt. Es wurden 174 mg (90% Reinheit, 8% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.09 min; MS (ESIpos): m/z = 246 [M+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 7.34-7.08 (m, 4H), 4.41 (dd, 1 H), 2.96 (dd, 1 H), 2.85 (dd, 1 H), 2.31 (s, 3H), 1.37 (s, 9H). Beispiel 33A
(+/-)-ie/f-Butyl-3-cyan-3-(2-methoxyphenyl)propanoat (Racemat)
Zu einer Lösung aus (2-Methoxyphenyl)acetonitril (5.00 g, 34.0 mmol) in THF (50 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (25 ml, 51 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde ie f-Butyl-bromacetat (7.5 ml, 51 mmol) langsam unter Rühren hinzu getropft. Das Kältebad wurde entfernt und das Gemisch unter Rühren über Nacht auf RT kommen gelassen. Anschließend wurde das Gemisch langsam mit Wasser versetzt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in einem Gemisch aus Acetonitril und Methanol (64 ml) gelöst und mittels prä- parativer HPLC [Säule: Kinetix C18, 5 μηι, 150 x 21.2 mm; Fluss: 30 ml/ min; Detektion: 210 nm; Injektionsvolumen: 0.5 ml; Eluent: 65% Wasser / 30% Acetonitril / 5% Ameisensäure (1 % in Wasser)— > 15% Wasser / 80% Acetonitril / 5% Ameisensäure (1 % in Wasser), Laufzeit: 6.0 min] gereinigt. Es wurden 4.90 g (96% Reinheit, 53% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.00 min; MS (ESIpos): m/z = 262 [M+H]+
Beispiel 34A
(+/-)-ie/f-Butyl-3-cyan-3-[2-(trifluormethoxy)phenyl]propanoat (Racemat)
Zu einer Lösung aus [2-(Trifluormethoxy)phenyl]acetonitril (5.00 g, 24.9 mmol) in THF (37 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (19 ml, 37 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde ie/f-Butyl-bromacetat (5.5 ml, 37 mmol) langsam unter Rühren hinzu getropft. Das Kältebad wurde entfernt und das Gemisch unter Rühren über Nacht auf RT kommen gelassen. Anschließend wurde das Gemisch langsam mit Wasser versetzt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels Flash-Säulenchromatographie (150 g Kieselgel, Cyclohe- xan— > Cyclohexan/Ethylacetat 95:5) gereinigt. Es wurden 6.70 g (65% Reinheit, 56% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.19 min; MS (ESIpos): m/z = 316 [M+H]+
Beispiel 35A
(+/-)-ie/f-Butyl-3-cyan-3-(pyridin-2-yl)propanoat (Racemat)
Zu einer Lösung aus Pyridin-2-ylacetonitril (5.00 g, 42.3 mmol) in THF (62 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (32 ml, 63 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde ie f-Butyl-bromacetat (9.4 ml, 63 mmol) langsam unter Rühren hinzu getropft. Das Kältebad wurde entfernt und das Gemisch unter Rühren über Nacht auf RT kommen gelassen. Anschließend wurde das Gemisch langsam mit Wasser versetzt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels präparativer HPLC [Säule: Kinetix C18, 5 μηη, 150 x 21.2 mm; Fluss: 30
ml/ min; Detektion: 210 nm; Injektionsvolumen: 0.75 ml; Eluent: 65% Wasser / 30% Acetonitril / 5% Ameisensäure (1 % in Wasser)— > 15% Wasser / 80% Acetonitril / 5% Ameisensäure (1 % in Wasser), Laufzeit: 6.0 min] gereinigt. Es wurden 1.94 g (98% Reinheit, 19% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.59 min; MS (ESIpos): m/z = 231 [M-H]" Beispiel 36A
(+/-)-ie/f-Butyl-3-cyan-3-[2-(trifluormethyl)phenyl]propanoat (Racemat)
Zu einer Lösung aus (Trifluormethyl)phenyl]acetonitril (2.50 g, 13.5 mmol) in THF (22 ml) unter Ar- gon wurde bei -78 langsam unter Rühren eine 2 M Lösung aus LDA in THF (10 ml, 20 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde ierf-Butyl-bromacetat (3.0 ml, 20 mmol) langsam unter Rühren bei -78 °C hinzu getropft. Das Gemisch wurde über Nacht weiter gerührt, wobei das Kältebad (Trocken- eis/Aceton) langsam auf RT kommen gelassen wurde. Anschließend wurde das Gemisch bei 0 °C mit Wasser und Ethylacetat (jeweils 50 ml) versetzt, geschüttelt und auf RT kommen gelassen. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (50 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung (100 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (50 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat Gradient 93:7 — > 7:3, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 2.7 g (98% Reinheit, 66% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.14 min; MS (ESIpos): m/z = 300 [M+H]+
Beispiel 37A
(+/-)-ie/f-Butyl-3-cyan-3-(3-methoxypyridin-2-yl)propanoat (Racemat)

Zu einer Lösung aus (3-Methoxypyridin-2-yl)acetonitril (2.00 g, 13.5 mmol, darstellbar gemäß US2003/220368 A1 , S. 30) in THF (20 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (10 ml, 20 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde ierf-Butyl-bromacetat (3.0 ml, 20 mmol) langsam unter Rühren hinzu getropft. Das Kältebad wurde entfernt und das Gemisch unter Rühren über Nacht auf RT kommen gelassen. Anschließend wurde das Gemisch langsam mit Wasser versetzt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels präparativer HPLC [Säule: Chromatorex C18, 10 μηι, 290 x 100 mm; Fluss: 250 ml/min; Detektion: 210 nm; Injektionsvolumen: 18 ml; Eluent: 70% Wasser / 30% Acetonitril -> 10% Wasser / 90% Acetonitril; Laufzeit: 40 min] gereinigt. Es wurden 2.32 g (99% Reinheit, 65% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.69 min; MS (ESIneg): m/z = 261 [M-H]-
Beispiel 38A
(+/-)-ie/f-Butyl-3-(2-chlorphenyl)-3-cyanbutanoat (Racen
Zu einer Lösung aus (+/-)-2-(2-Chlorphenyl)propannitril (5.00 g, 30.2 mmol, darstellbar gemäß ChemCatChem 2014, 6 (8), 2425-2431 ) in THF (42 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (23 ml, 45 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde ie/f-Butyl-bromacetat (5.3 ml, 36 mmol) langsam unter Rühren hinzu getropft. Das Kältebad wurde entfernt und das Gemisch unter Rühren über Nacht auf RT kommen gelassen. Anschließend wurde das Gemisch langsam mit Wasser versetzt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat 8:2) gereinigt. Es wurden 6.30 g (97% Reinheit, 72% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.1 1 min; MS (ESIpos): m/z = 280 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.256 (16.00), 1.390 (0.79), 1.397 (4.13), 1.889 (4.42), 3.1 16 (0.68), 3.155 (1.02), 3.297 (1.02), 3.313 (0.96), 3.336 (0.68), 7.406 (0.44), 7.410 (0.66), 7.420 (0.95), 7.429 (0.88), 7.434 (0.57), 7.525 (0.55), 7.564 (0.55).
Beispiel 39A
(+/-)-fe/f-Butyl-3-(2-chlor-6-fluorphenyl)-3-cyanbutanoat (Racemat)
Zu einer Lösung aus (+/-)-2-(2-Chlor-6-fluorphenyl)propannitril (5.00 g, 27.2 mmol), darstellbar aus (2-Chlor-6-fluorphenyl)acetonitril durch Methylierung mit Tetrabutylammonium-Iodid und Ka- liumcarbonat in Toluol bei 80°C (analog zu ChemCatChem 2014, 6 (8), 2425-2431 ) in THF (38 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (20 ml, 41 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde ie/f-Butyl-bromacetat (4.8 ml, 33 mmol) langsam unter Rühren hinzu getropft. Das Kältebad wurde entfernt und das Gemisch unter Rühren über Nacht auf RT kommen gelassen. Anschließend wurde das Gemisch langsam mit Wasser ver- setzt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash- Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohe- xan/Ethylacetat 8:2) gereinigt. Es wurden 5.26 g (98% Reinheit, 64% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.12 min; MS (ESIpos): m/z = 298 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.298 (16.00), 1.862 (2.46), 1.868 (2.44), 3.246 (0.50), 3.259 (0.51 ), 3.31 1 (0.83), 3.406 (0.53), 3.415 (0.52), 7.412 (1.35), 7.419 (0.70), 7.422 (0.55), 7.429 (0.41 ). Beispiel 40A
(+/-)-ie/f-Butyl-3-cyan-3-(2-methoxyphenyl)butanoat (Racemat)
Zu einer Lösung aus 2-(2-Methoxyphenyl)propannitril (8.00 g, 49.6 mmol, darstellbar gemäß Or- ganic Letters 2008, 10 (20), 4573-4576) in THF (73 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (37 ml, 74 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde ie/f-Butyl-bromacetat (1 1 ml, 74 mmol) langsam unter Rühren hinzu getropft. Das Kältebad wurde entfernt und das Gemisch unter Rühren über Nacht auf RT kommen gelassen. Anschließend wurde das Gemisch langsam mit Wasser versetzt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewa- sehen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels präparativer HPLC [Säule: Chromatorex C18, 10 μηι, 370 x 100 mm; Fluss: 250 ml/min; Detekti- on: 210 nm; Temperatur: 22 °C; Injektionsvolumen: 18 ml; Eluent: 30% Wasser / 70% Acetonitril -> 10% Wasser / 90% Acetonitril; Laufzeit: 31 min] gereinigt. Es wurden 7.93 g (95% Reinheit, 55% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.05 min; MS (ESIpos): m/z = 276 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.215 (16.00), 1.782 (4.55), 2.949 (0.80), 2.987 (1.16), 3.131 (1.14), 3.169 (0.78), 3.312 (0.57), 6.989 (0.56), 6.992 (0.59), 7.098 (0.52), 7.1 17 (0.61 ), 7.360 (0.49), 7.370 (0.67), 7.375 (0.63), 7.390 (0.59), 7.394 (0.43).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 7.41 -7.31 (m, 2H), 7.1 1 (d, 1 H), 6.99 (td, 1 H), 3.86 (s, 3H), 3.15 (d, 1 H), 2.97 (d, 1 H), 1.78 (s, 3H), 1.21 (s, 9H).
Beispiel 41A
(+/-)-ie/f-Butyl-3-cyan-3-(5-fluor-2-methoxyphenyl)propanoat (Racemat)
Zu einer Lösung aus Lithium-bis(trimethylsilyl)amid (6.36 ml, 1 M, 6.36 mmol) in THF (22 ml) un ter Stickstoff wurde bei -78 °C tropfenweise eine Lösung aus (5-Fluor-2-methoxyphenyl) acetonitril (1.00 g, 6.05 mmol) in THF (22 ml) hinzugegeben. Nach 45 min Rühren bei -78 °C
wurde das Reaktionsgemisch mit einer Lösung aus ie f-Butyl-bromacetat (1.18 g, 6.05 mmol) in THF (22 ml) versetzt und eine weitere Stunde bei -78 °C, gefolgt von 2 h bei RT gerührt. Anschließend wurde das Gemisch mit Ethylacetat und Wasser versetzt, geschüttelt, und die Phasen wurden getrennt. Die wässrige Phase wurde zweimal mit Ethylacetat extrahiert. Die vereinig- ten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde mittels Flash- Säulenchromatographie (40 g Kieselgel Reveleris, Heptan/Ethylacetat-Gradient) vorgereinigt. Das vorgereinigte Produkt wurde anschließend mittels Flash-Säulenchromatographie (40 g Re- versed Phase C18 Reveleris, Fluss 40 ml/min, (Wasser + 0.1 % Ameisensäure )/( Aceton itril + 0.1 % Ameisensäure)-Gradient) nachgereinigt. Es wurden 941 mg (Reinheit 98%, 55% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 10): Rt = 2.28 min; MS (ESpos): m/z = 297 [M]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 7.15 (d, 1 H), 7.02 (dd, 1 H), 6.84 (dd, 1 H), 4.47 (dd, 1 H), 3.86 (s, 3H), 2.90-2.70 (m, 2H), 1.45 (s, 9H).
Beispiel 42A
(+/-)-ie/f-Butyl-3-cyan-3-(2-fluor-6-methoxyphenyl)propanoat (Racen
Zu einer Lösung aus (2-Fluor-6-methoxyphenyl)acetonitril (5.00 g, 30.3 mmol) in THF (50 ml) unter Argon wurde bei -78 bis -60 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (23 ml, 45 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde ie f-Butyl-bromacetat (6.7 ml, 45 mmol) langsam unter Rühren bei -78 bis -60 °C hinzu getropft. Das Kältebad wurde entfernt und das Gemisch unter Rühren langsam auf RT kommen gelassen. Nach 4 h wurde das Gemisch bei 0 °C mit Wasser und Ethylacetat (jeweils 50 ml) versetzt, geschüttelt, und die Phasen wurden getrennt. Die wässrige Phase wurde einmal mit Ethylacetat (50 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung (100 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 1 1.4 g („93% Reinheit",„>100% d. Th.", weitere Verunreinigungen enthalten) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.00 min; MS (ESIpos): m/z = 280 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.334 (16.00), 1.397 (0.78), 1.425 (3.42), 2.844 (0.47), 2.862 (0.47), 2.999 (0.42), 3.020 (0.42), 3.309 (3.17), 4.017 (0.76), 4.635 (0.44), 6.894 (0.43), 6.958 (0.53), 6.980 (0.58), 7.402 (0.46), 7.419 (0.45).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 7.41 (dd, 1 H), 6.97 (d, 1 H), 6.89 (t, 1 H), 4.63 (dd, 1 H), 3.89 (s, 3H), 3.03 (dd, 1 H), 2.83 (dd, 1 H), 1.33 (s, 9H).
Beispiel 43A
(+/-)-ie/f-Butyl-3-cyan-3-(2-ethoxyphenyl)propanoat (Racemat)
Zu einer Lösung aus Diisopropylamin (2.9 ml, 20 mmol) in THF (1 10 ml) wurde bei -78 °C trop- fenweise eine Lösung von 1.6 M Butyllithium in Hexan (13 ml, 20 mmol) gegeben, und das Gemisch wurde 45 min bei -78 °C gerührt. Anschließend wurde eine Lösung aus (2-Ethoxyphenyl)acetonitril (3.00 g, 18.6 mmol) in THF (1 10 ml) tropfenweise hinzugegeben. Nach einer Stunde Rühren bei -78 °C wurde das Gemisch mit einer Lösung aus ierf-Butyl- bromacetat (2.8 ml, 20 mmol) in THF (15 ml) versetzt und weitere 45 min bei -78 °C gerührt. An- schließend wurde das Kältebad entfernt, und das Gemisch wurde weitere 2 h bei RT gerührt. Nach erneutem Abkühlen auf -78 °C wurde das Gemisch mit gesättigter Ammoniumchlorid- Lösung versetzt, auf RT kommen gelassen und mit Ethylacetat versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde mittels Flash-Säulenchromatographie (40 g Reversed Phase C18 Reveleris, Fluss 100 ml/min, (Wasser + 0.1 % Ameisensäure )/(Acetonitril + 0.1 % Ameisensäure)-Gradient) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 1.96 g (99% Reinheit, 38% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 10): Rt = 2.22 min; MS (ESpos): m/z = 293 [M+18]+
1H-NMR (400 MHz, CDCI3): δ [ppm] = 7.39 (d, 1 H), 7.38-7.25 (m, teilweise verdeckt, 1 H), 7.00- 6.90 (m, 1 H), 6.88 (d, 1 H), 4.50 (dd, 1 H), 4.10 (dd, 2H), 2.90-2.70 (m, 2H), 1.50-1.43 (m, 12H).
Beispiel 44A
(+/-)-ie/f-Butyl-3-cyan-3-(5-fluor-2-methoxyphenyl)butanoat (Racemat)
Zu einer Lösung aus Diisopropylamin (2.3 ml, 16 mmol) in THF (80 ml) unter Stickstoff wurde bei -78 °C tropfenweise eine Lösung von 1.6 M Butyllithium in Hexan (10 ml, 16 mmol) gegeben, und das Gemisch wurde 45 min bei -78 °C gerührt. Anschließend wurde eine Lösung aus (+/-)-2- (5-Fluor-2-methoxyphenyl)propannitril (2.40 g, 13.4 mmol, Beispiel 6A) in THF (80 ml) tropfenweise hinzugegeben. Nach 50 min Rühren bei -78 °C wurde das Gemisch mit einer Lösung aus ie/f-Butyl-bromacetat (2.3 ml, 16 mmol) in THF (10 ml) versetzt und weitere 30 min bei -78 °C gerührt. Anschließend wurde das Kältebad entfernt, und das Gemisch wurde weitere 2 h bei RT gerührt. Das Gemisch wurde mit gesättigter Ammoniumchlorid-Lösung versetzt und geschüttelt. Nach Zugabe von Ethylacetat wurden die Phasen getrennt und die wässrige Phase zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde mittels Flash-Säulenchromatographie (120 g Kieselgel Reveleris, Fluss 80 ml/min, Heptan/Dichlormethan-Gradient 1 :0 — > 0:1 , Laufzeit 38 min) vorgereinigt. Es wurden zwei vorgereinigte Produkt-Fraktionen erhalten, welche mittels Flash-Säulenchromatographie (40 g bzw. 80 g Kieselgel Reveleris, Fluss 40 ml/min bzw. 55 ml/min, Heptan/Ethylacetat- Gradient 1 :0— > 7: bzw. 31 :0— > 6:4, Laufzeit 32 min bzw. 25 min) nachgereinigt wurden. Es wurden 486 mg (97% Reinheit, 12% d. Th., siehe Analytik) einer ersten Charge und 890 mg (96% Reinheit, 22% d. Th.) einer zweiten Charge der Titelverbindung erhalten.
LC-MS (Methode 10): Rt = 2.27 min; MS (ESpos): m/z = 294 [M+1]+
GC-MS (Methode 13): Rt = 3.84 min; MS (ESIpos): m/z = 293 [M]+
1H-NMR (400 MHz, CDCI3): δ [ppm] = 7.24 (d, 1 H), 7.05-6.93 (m 1 H), 7.90-7.81 (m, 1 H), 3.89 (s, 3H), 3.18 (d, 1 H), 2.96 (d, 1 H), 1.86 (s, 3H), 1.31 (s, 9H).
Beispiel 45A
(+/-)-ie/f-Butyl-3-cyan-3-(3,5-difluor-2-methoxyphenyl)butanoat (Racemat)

Zu einer Lösung aus Diisopropylamin (1.8 ml, 13 mmol) in THF (25 ml) unter Stickstoff wurde bei -78 °C tropfenweise eine Lösung von 1.6 M Butyllithium in Hexan (7.6 ml, 12 mmol) gegeben, und das Gemisch wurde 30 min bei -78 °C gerührt. Anschließend wurde eine Lösung aus (+/-)-2- (3,5-Difluor-2-methoxyphenyl)propannitril (1.99 g, 10.1 mmol, Beispiel 7A) in THF (10 ml) trop- fenweise hinzugegeben. Nach 30 min Rühren bei -78 °C wurde das Gemisch mit einer Lösung aus ie/f-Butyl-bromacetat (2.56 g, 13.1 mmol) in THF (25 ml) versetzt und weitere 30 min bei -78 °C gerührt. Anschließend wurde das Gemisch mit gesättigter Ammoniumchlorid-Lösung versetzt, geschüttelt und auf RT kommen gelassen. Durch Zugabe von 1 M Salzsäure wurde die wässrige Phase auf ca. pH 3 eingestellt. Nach Zugabe von gesättigter Natriumchlorid-Lösung wurde das Gemisch dreimal mit Ethylacetat extrahiert, und die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels Flash-Säulenchromatographie (120 g Kieselgel Reveleris, Fluss 40 ml/min, Laufmittel Dichlormethan) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 1.54 g (99% Reinheit, 49% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 9): Rt = 2.26 min; MS (ESpos): m/z = 312 [M+1]+
GC-MS (Methode 13): Rt = 3.64 min; MS (ESIpos): m/z = 31 1 [M]+
Beispiel 46A
(+/-)-ie/f-Butyl-3-cyan-3-(4-fluor-2-methoxyphenyl)butanoat (Racemat)

Zu einer Lösung aus Diisopropylamin (3.7 ml, 27 mmol) in THF (50 ml) unter Stickstoff wurde bei -78 °C tropfenweise eine Lösung von 1.6 M Butyllithium in Hexan (16 ml, 25 mmol) gegeben, und das Gemisch wurde 30 min bei -78 °C gerührt. Anschließend wurde eine Lösung aus (+/-)-2- (4-Fluor-2-methoxyphenyl)propannitril (3.77 g, 21.0 mmol, Beispiel 8A) in THF (20 ml) tropfenweise hinzugegeben. Nach 30 min Rühren bei -78 °C wurde das Gemisch mit einer Lösung aus ie/f-Butyl-bromacetat (5.33 g, 27.3 mmol) in THF (50 ml) versetzt und weitere 30 min bei -78 °C gerührt. Anschließend wurde das Gemisch mit Wasser versetzt, geschüttelt und auf RT kommen gelassen. Durch Zugabe von 1 M Salzsäure wurde die wässrige Phase auf ca. pH 3 eingestellt. Nach Zugabe von gesättigter Natriumchlorid-Lösung wurde das Gemisch dreimal mit Ethylacetat extrahiert, und die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels Flash-Säulenchromatographie (220 g Kieselgel,
Laufmittel Dichlormethan, Fluss 80 ml/min) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 3.02 g (99% Reinheit, 49% d. Th.) der Titelverbindung erhalten.
GC-MS (Methode 13): Rt = 3.81 min; MS (ESIpos): m/z = 293 [M]+
1H-NMR (400 MHz, CDCI3): δ [ppm] = 7.50-7.40 (m, 1 H), 6.70-6.65 (m, 2H), 3.89 (s, 3H), 3.14 (d, 1 H), 2.95 (d, 1 H), 1.86 (s, 3H), 1.30 (s, 9H).
Beispiel 47A
(+/-)-ie/f-Butyl-3-cyan-3-(2-fluor-6-methoxyphenyl)butanoat (Racemat)

Zu einer Lösung aus (+/-)-2-(2-Fluor-6-methoxyphenyl)propannitril (578 mg, 3.22 mmol, Beispiel 9A) in THF (6.0 ml) unter Argon wurde bei -78 langsam unter Rühren eine 2 M Lösung aus LDA in THF (2.4 ml, 4.8 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde ierf-Butyl-bromacetat (943 mg, 4.84 mmol) langsam unter Rühren bei -78 °C hinzu getropft. Nach 3 h Rühren bei -78 °C wurde das Gemisch mit Wasser und Ethylacetat (jeweils 50 ml) versetzt, geschüttelt und auf RT kommen gelassen. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (50 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung (100 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (50 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat Gradient 93:7 — > 7:3, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 809 mg (80% Reinheit, 68% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.01 min; MS (ESIpos): m/z = 294 [M+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 7.39-7.30 (m, 1 H), 6.98-6.93 (m, 1 H), 6.85-6.74 (m, 1 H), 3.85 (s, 3H), 3.27 (dd, 1 H), 3.05 (dd, 1 H), 1.76 (d, 3H), 1.27 (s, 9H).
Beispiel 48A
ie/f-Butyl-3-(2-chlorphenyl)-4-nitropentanoat (Diastereomerengemisch)
Zu einer Lösung aus ie f-Butyl-(2£)-3-(2-chlorphenyl)acrylat (1.00 g, 4.19 mmol, darstellbar gemäß Beilstein Journal of Organic Chemistry 2QW, 8, 1747-1752) in Nitroethan (8.0 ml, 92 mmol) wurde 2-ie/f-Butyl-1 ,1 ,3,3-tetramethylguanidin (210 μΙ, 1.0 mmol) hinzugegeben, und das Ge- misch wurde über Nacht unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das Gemisch filtriert und mittels präparativer HPLC (Methode 16) gereinigt. Es wurden 1.10 g (Reinheit ca. 90% laut Ή-NMR, 75% d. Th.) der Titelverbindung erhalten.
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.153 (16.00), 1.180 (12.81 ), 1.266 (2.26), 1.283 (2.28), 1.302 (0.50), 1.308 (1.93), 1.504 (1.82), 1.520 (1.84), 2.030 (0.60), 2.644 (0.50), 2.656 (0.49), 2.720 (0.45), 2.745 (0.52), 2.751 (0.51 ), 2.766 (0.56), 2.771 (0.51 ), 2.798 (0.47), 7.290 (0.42), 7.294 (0.44), 7.309 (0.44), 7.314 (0.59), 7.328 (0.52), 7.333 (0.73), 7.338 (0.64), 7.344 (0.43), 7.358 (0.50), 7.362 (0.50), 7.410 (0.42), 7.414 (0.41 ), 7.480 (0.56), 7.483 (0.59), 7.492 (0.82), 7.498 (0.86), 7.502 (0.47), 7.51 1 (0.49), 7.516 (0.41 ).
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 7.70-6.93 (m, 4H), 5.22-4.81 (m, 1 H), 4.27-3.94 (m, 1 H), 2.87-2.57 (m, 2H), 1.51 & 1.27 (2x d, 3H), 1.18 & 1.15 (2x d, 9H).
Beispiel 49A
(+/-)-ie/f-Butyl-4-(2-chlorphenyl)-4-cyanbutanoat (Racemat)
Zu einer Lösung aus (2-Chlorphenyl)acetonitril (5.00 g, 33.0 mmol) in THF (46 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (25 ml, 49 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde ief-Butyl-3-brompropanoat (8.28 g, 39.6 mmol) langsam unter Rühren bei -78 °C hinzu getropft. Das Gemisch wurde über Nacht weiter gerührt, wobei das Kältebad (Trockeneis/Aceton) langsam auf RT kommen gelassen wurde. Anschließend wurde das Gemisch langsam mit Wasser versetzt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natri-
umsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde mittels präparativer HPLC (Methode 19) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 5.20 g (75% Reinheit, 42% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.22 min; MS (ESIpos): m/z = 280 [M+H]+ Beispiel 50A
(+/-)-ie/f-Butyl-4-(2-chlorphenyl)-4-cyanpentanoat (Racemat)
Zu einer Lösung aus 2-(2-Chlorphenyl)propannitril (5.00 g, 30.2 mmol, darstellbar durch Methylie- rung von 2-(2-Chlorphenyl)acetonitril gemäß WO2011/123419 A1 , S. 386-387) in THF (42 ml) un- ter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (23 ml, 45 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde ierf-Butyl-3-brompropanoat (7.57 g, 36.2 mmol) langsam unter Rühren bei -78 °C hinzu getropft. Das Gemisch wurde über Nacht weiter gerührt, wobei das Kältebad (Trockeneis/Aceton) langsam auf RT kommen gelassen wurde. Anschließend wurde das Ge- misch langsam mit Wasser versetzt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde mittels präparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 2.21 g (81% Reinheit, 20% d. Th.) der Titelverbindung erhalten. LC-MS (Methode 1 ): Rt = 2.27 min; MS (ESIpos): m/z = 294 [M+H]+
Beispiel 51A
(+/-)-ie/f-Butyl-4-(2-chlor-6-fluorphenyl)-4-cyanbutanoat (Racemat)
Zu einer Lösung aus (2-Chlor-6-fluorphenyl)acetonitril (2.00 g, 11.8 mmol) in THF (15 ml) unter Ar- gon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (8.8 ml, 18 mmol)
gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde ief-Butyl-3-brompropanoat (2.2 ml, 14 mmol) langsam unter Rühren bei -78 °C hinzu getropft. Das Gemisch wurde über Nacht weiter gerührt, wobei das Kältebad (Trockeneis/Aceton) langsam auf RT kommen gelassen wurde. Anschließend wurde das Gemisch langsam mit Wasser versetzt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde mittels Flash-Säulenchromatographie (50 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat-Gradient 93:7— > 6:4, Isolera One) vorgereinigt. Anschließend wurde mittels präparativer HPLC (Methode 16) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 1.45 g (100% Reinheit, 41% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.16 min; MS (ESIpos): m/z = 298 [M+H]+ Beispiel 52A
(+/-)-ie/f-Butyl-4-cyan-4-(2-methylphenyl)butanoat (Racemat)
Zu einer Lösung aus (2-Methylphenyl)acetonitril (2.00 g, 15.2 mmol) in THF (17 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (1 1 ml, 23 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde ierf-Butyl-3-brompropanoat (2.9 ml, 18 mmol) langsam unter Rühren bei -78 °C hinzu getropft. Das Gemisch wurde über Nacht weiter gerührt, wobei das Kältebad (Trockeneis/Aceton) langsam auf RT kommen gelassen wurde. Anschließend wurde das Gemisch langsam mit Wasser versetzt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde mittels Flash-Säulen- Chromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat 85:15, Isolera One) vorgereinigt. Anschließend wurde mittels präparativer HPLC (Methode 16) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 534 mg (98% Reinheit, 13% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.17 min; MS (ESIpos): m/z = 260 [M+H]+
Beispiel 53A
(+/-)-fe/f-Butyl-4-(2-chlor-5-fluorphenyl)-4-cyanbutanoat (Racemat)
Zu einer Lösung aus (2-Chlor-5-fluorphenyl)acetonitril (10.0 g, 59.0 mmol) in THF (75 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (44 ml, 88 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf - 78 °C abgekühlt. Anschließend wurde ie f-Butyl-3-brompropanoat (1 1 ml, 71 mmol) langsam unter Rühren bei -78 °C hinzu getropft. Das Gemisch wurde über Nacht weiter gerührt, wobei das Kältebad (Trockeneis/Aceton) langsam auf RT kommen gelassen wurde. Anschließend wurde das Gemisch langsam mit Wasser versetzt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde mittels Flash-Säulenchromatographie (340 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohe- xan/Ethylacetat-Gradient 93:7— > 3:7, Isolera One) gereinigt. Die vereinigten Zielfraktionen wur- den eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 14.8 g (93% Reinheit, 78% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.22 min; MS (ESIpos): m/z = 298 [M+H]+ Beispiel 54A
(+/-)-ie/f-Butyl-4-cyan-4-(2-methoxyphenyl)butanoat (Racemat)
Zu einer Lösung aus (2-Methoxyphenyl)acetonitril (2.00 g, 13.6 mmol) in THF (17 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (10 ml, 20 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde ie f-Butyl-3-brompropanoat (2.6 ml, 16 mmol) langsam unter Rühren bei -78 °C hinzu getropft. Das Gemisch wurde über Nacht weiter gerührt, wobei das Kältebad (Trockeneis/Aceton) langsam auf RT kommen gelassen wurde. Anschließend wurde das
Gemisch langsam mit Wasser versetzt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohe- xan/Ethylacetat-Gradient 93:7— > 6:4, Isolera One) vorgereinigt. Anschließend wurde mittels prä- parativer HPLC (Methode 16) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 495 mg (98% Reinheit, 13% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.13 min; MS (ESIpos): m/z = 276 [M+H]+ Beispiel 55A
(+/-)-ie/f-Butyl-4-(2-chlor-5-fluorphenyl)-4-cyanpentanoat (Racemat)
Zu einer Lösung aus (+/-)-2-(2-Chlor-5-fluorphenyl)propannitril (2.50 g, 13.6 mmol, Beispiel 10A) in THF (17 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (10 ml, 20 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde ie f-Butyl-3-brompropanoat (2.6 ml, 16 mmol) langsam unter Rühren bei -78 °C hinzu getropft. Das Gemisch wurde über Nacht weiter gerührt, wobei das Kältebad (Trockeneis/Aceton) langsam auf RT kommen gelassen wurde. Anschließend wurde das Gemisch langsam mit Wasser versetzt und zweimal mit Ethylacetat extra- hiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde mittels Flash-Säulenchromatographie (340 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat-Gradient 93:7— > 3:7, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 1.20 g (75% Rein- heit, 21 % d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.29 min; MS (ESIpos): m/z = 312 [M+H]+
Beispiel 56A
(+/-)-ie/f-Butyl-4-(2-chlor-3,6-difluorphenyl)-4-cyanbutanoat (Racemat)
Zu einer Lösung aus (2-Chlor-3,6-difluorphenyl)acetonitril (3.84 g, 20.5 mmol, Beispiel 1 1A) in THF (15 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (12 ml, 25 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde eine Lösung aus ie/f-Butyl-3- brompropanoat (2.6 ml, 16 mmol) in THF (10 ml) langsam unter Rühren bei -78 °C hinzu getropft. Das Gemisch wurde über Nacht weiter gerührt, wobei das Kältebad (Trockeneis/Aceton) langsam auf RT kommen gelassen wurde. Anschließend wurde das Gemisch bei ca. 0 °C langsam mit Wasser und Ethylacetat (jeweils 100 ml) versetzt und geschüttelt. Nach Phasentren- nung wurde die wässrige Phase einmal mit Ethylacetat (100 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung (150 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dich- lormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge Ultra, Cyclohexan/Ethylacetat-Gradient 93:7— > 7:3, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 4.14 g (95% Reinheit, 61 % d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.17 min; MS (ESIpos): m/z = 338 [M+Na]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 7.58 (td, 1 H), 7.45 (td, 1 H), 4.68 (t, 1 H), 2.39-2.32 (m, 2H), 2.29-2.17 (m, 1 H), 2.17-2.05 (m, 1 H), 1.37 (s, 9H). Beispiel 57A
(+/-)-ie/f-Butyl-4-cyan-4-(2-fluorphenyl)butanoat (Racemat)
Zu einer Lösung aus (2-Fluorphenyl)acetonitril (5.00 g, 37.0 mmol) in THF (35 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (22 ml, 44 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde eine Lösung aus ie f-Butyl-3-brompropanoat (7.0 ml, 44 mmol) in
THF (10 ml) langsam unter Rühren bei -78 °C hinzu getropft. Das Gemisch wurde über Nacht weiter gerührt, wobei das Kältebad (Trockeneis/Aceton) langsam auf RT kommen gelassen wurde. Anschließend wurde das Gemisch bei ca. 0 °C langsam mit Wasser und Ethylacetat (jeweils 100 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (100 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung (150 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash- Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge Ultra, Cyclohexan/ Ethylacetat- Gradient 93:7— > 7:3, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 4.22 g (69% Reinheit, 29% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.08 min; MS (ESIpos): m/z = 264 [M+H]+ Beispiel 58A
(+/-)-ie/f-Butyl-4-cyan-4-[2-(difluormethoxy)phenyl]butanoat (Racemat)
Zu einer Lösung aus [2-(Difluormethoxy)phenyl]acetonitril (5.00 g, 27.3 mmol) in THF (25 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (16 ml, 33 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf - 78 °C abgekühlt. Anschließend wurde eine Lösung aus ie f-Butyl-3-brompropanoat (5.2 ml, 33 mmol) in THF (10 ml) langsam unter Rühren bei -78 °C hinzu getropft. Das Gemisch wurde über Nacht weiter gerührt, wobei das Kältebad (Trockeneis/Aceton) langsam auf RT kommen gelassen wurde. Anschließend wurde das Gemisch bei ca. 0 °C langsam mit Wasser und Ethylacetat (jeweils 100 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (100 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung (150 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge Ultra, Cyclohe- xan/Ethylacetat-Gradient 93:7— > 7:3, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 3.85 g (83% Reinheit, 38% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.10 min; MS (ESIpos): m/z = 312 [M+H]+
Beispiel 59A
(+/-)-fe/f-Butyl-4-cyan-4-(2,6-difluorphenyl)butanoat (Racemat)
Zu einer Lösung aus (2,6-Difluorphenyl)acetonitril (5.00 g, 32.7 mmol) in THF (30 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (20 ml, 39 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde eine Lösung aus ief-Butyl-3-brompropanoat (6.2 ml, 39 mmol) in THF (10 ml) langsam unter Rühren bei -78 °C hinzu getropft. Das Gemisch wurde über Nacht weiter gerührt, wobei das Kältebad (Trockeneis/Aceton) langsam auf RT kommen gelassen wurde. An- schließend wurde das Gemisch bei ca. 0 °C langsam mit Wasser und Ethylacetat (jeweils 100 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (100 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung (150 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromato- graphie (100 g Kieselgel Biotage Snap-Cartridge Ultra, Cyclohexan/Ethylacetat-Gradient 93:7— > 7:3, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 4.87 g (76% Reinheit, 40% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.06 min; MS (ESIpos): m/z = 282 [M+H]+
Beispiel 60A
(+/-)-ie/f-Butyl-4-(2-chlor-3-fluorphenyl)-4-cyanbutanoat (Racemat)
Zu einer Lösung aus (2-Chlor-3-fluorphenyl)acetonitril (4.00 g, 23.6 mmol, Beispiel 12A) in THF (30 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (14 ml, 28 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf - 78 °C abgekühlt. Anschließend wurde eine Lösung aus ierf-Butyl-3-brompropanoat (4.5 ml, 28 mmol) in THF (20 ml) langsam unter Rühren bei -78 °C hinzu getropft. Das Gemisch wurde über
Nacht weiter gerührt, wobei das Kältebad (Trockeneis/Aceton) langsam auf RT kommen gelassen wurde. Anschließend wurde das Gemisch bei ca. 0 °C langsam mit Wasser und Ethylacetat (jeweils 100 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (100 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesät- tigter wässriger Natriumchlorid-Lösung (150 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash- Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge Ultra, Cyclohexan/Ethylacetat- Gradient 93:7— > 7:3, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 4.36 g (95% Reinheit, 59% d. Th.) der Titelver- bindung erhalten.
LC-MS (Methode 1 ): Rt = 2.24 min; MS (ESIpos): m/z = 298 [M+H]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.007 (0.14), 1.174 (0.02), 1.222 (0.06), 1.342 (0.84), 1.346 (0.35), 1.382 (16.00), 1.429 (0.05), 1.539 (0.06), 1.987 (0.03), 2.071 (0.04), 2.090 (0.09), 2.106 (0.20), 2.125 (0.45), 2.142 (0.53), 2.162 (0.44), 2.182 (0.23), 2.196 (0.08), 2.217 (0.06), 2.252 (0.02), 2.302 (0.07), 2.317 (0.08), 2.343 (0.38), 2.358 (0.60), 2.362 (0.45), 2.376 (0.75), 2.395 (0.29), 2.417 (0.12), 2.436 (0.04), 2.669 (0.03), 2.709 (0.02), 3.730 (0.02), 4.174 (0.10), 4.566 (0.34), 4.586 (0.43), 4.603 (0.33), 7.426 (0.24), 7.431 (0.29), 7.444 (0.48), 7.449 (0.60), 7.460 (0.54), 7.466 (0.30), 7.476 (0.47), 7.482 (0.52), 7.491 (0.55), 7.508 (0.28), 7.529 (0.09).
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 7.55-7.40 (m, 3H), 4.59 (dd, 1 H), 2.45-2.29 (m, 2H), 2.23-2.06 (m, 2H), 1.38 (s, 9H).
Beispiel 61A
(+/-)-ie/f-Butyl-/V-[(2-chlorphenyl)(cyan)methyl]-/V-methylglycinat (Racemat)
Eine Lösung aus ie/f-Butyl-/V-methylglycinat-Hydrochlorid (1.03 g, 7.1 1 mmol) in Ethylacetat (10 ml) wurde mit gesättigter Natriumcarbonat-Lösung versetzt und geschüttelt. Nach Phasentrennung wurde die organische Phase über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in Dichlormethan (50 ml) gelöst und tropfenweise mit 2-Chlorbenzaldehyd (1.00 g, 7.1 1 mmol) bei RT versetzt. Nach 15 min Rühren bei RT wurde Trimethylsilancarbonitril (706 mg, 7.1 1 mmol) tropfenweise hinzugegeben, und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt, und der Rückstand wurde in Ethylacetat aufgenommen und mit gesättigter wässriger Natriumchlorid-Lösung gewaschen. Die organische
Phase wurde über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde mittels präparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 1.47 g (90% Reinheit, 63% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.19 min; MS (ESIpos): m/z = 295 [M+H]+ Beispiel 62A
(+/-)-ie/f-Butyl-/V-[(2-chlorphenyl)(cyan)methyl]glycinat (Racemat)
Zu einer Lösung aus ie/f-Butylglycinat (933 mg, 7.1 1 mmol) in Dichlormethan (50 ml) wurde trop- fenweise 2-Chlorbenzaldehyd (1.00 g, 7.1 1 mmol) bei RT gegeben. Nach 15 min Rühren bei RT wurde Trimethylsilancarbonitril (706 mg, 7.1 1 mmol) tropfenweise hinzugegeben, und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt, und der Rückstand wurde in Ethylacetat aufgenommen und mit gesättigter wässriger Natriumchlorid- Lösung gewaschen. Die organische Phase wurde über Natriumsulfat getrocknet, filtriert und ein- geengt, und der Rückstand wurde mittels präparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 1.47 g (Reinheit 33%, 24 % d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.01 min; MS (ESIpos): m/z = 225 [M-C4H8+H]+
Beispiel 63A
(+/-)-Methyl-{[1 -(2-chlorphenyl)-2-nitroethyl]sulfanyl}acetat (Racemat)
Eine Lösung aus 1 -Chlor-2-[(£)-2-nitrovinyl]benzol (1.00 g, 5.45 mmol) in Dichlormethan (50 ml) unter Argon wurde bei -30 °C tropfenweise mit einer Lösung aus Methyl-sulfanylacetat (578 mg, 5.45 mmol) in Dichlormethan (25 ml) versetzt, und das Gemisch wurde 2 h bei -30 °C und 16 h bei RT gerührt. Anschließend wurde das Gemisch eingeengt, und der Rückstand wurde in Ace-
tonitril gelöst und mittels präparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 1.52 g (100% Reinheit, 97% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 0.96 min; MS (ESIpos): m/z = 307
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 7.62-7.59 (m, 1 H), 7.52-7.48 (m, 1 H), 7.40-7.32 (m, 2H), 5.35 (dd, 1 H), 5.29 (dd, 1 H), 5.13 (dd, 1 H), 3.60 (s, 3H), 3.59 (d, 1 H), 3.43 (d, 1 H).
Beispiel 64A
(+/-)-ie/f-Butyl-4-(6-chlor-2,3-difluorphenyl)-4-cyanbutanoat (Racemat)
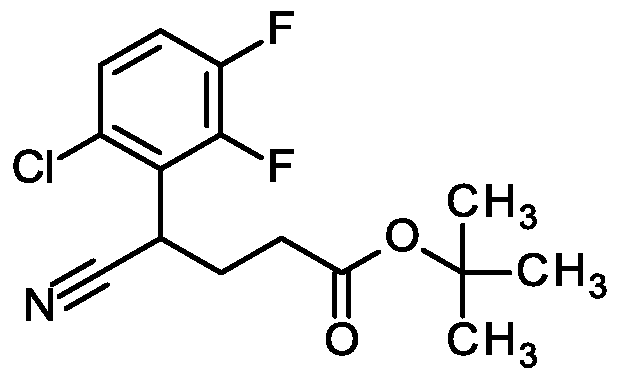
Zu einer Lösung aus (6-Chlor-2,3-difluorphenyl)acetonitril (2.23 g, 1 1.9 mmol, nicht reinheitskor- rigiert, Beispiel 13A) in THF (15 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (7.1 ml, 14 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde eine Lösung aus ie f-Butyl-3-brompropanoat (4.5 ml, 28 mmol) in THF (10 ml) langsam unter Rühren bei -78 °C hinzu getropft. Das Gemisch wurde über Nacht weiter gerührt, wobei das Kältebad (Trocken- eis/Aceton) langsam auf RT kommen gelassen wurde. Anschließend wurde das Gemisch bei ca. 0 °C langsam mit Wasser (50 ml) und Ethylacetat (100 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (50 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung (80 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (50 g Kieselgel Biotage Snap-Cartridge Ultra, Cyclohexan/Ethylacetat-Gradient 93:7— > 7:3, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 2.29 g (83% Reinheit, 51 % d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.17 min; MS (ESIpos): m/z = 316 [M+H]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 7.73-7.53 (m, 1 H), 7.52-7.41 (m, 1 H), 4.68 (t, 1 H), 2.43- 2.32 (m, 2H), 2.32-2.18 (m, 1 H), 2.18-2.04 (m, 1 H), 1.37 (s, 9H).
Beispiel 65A
(+/-)-Methyl-4-cyan-4-[2-(trifluormethyl)phenyl]butanoat (Racemat)
Zu einer Lösung aus [2-(Trifluormethyl)phenyl]acetonitril (5.00 g, 27.0 mmol) in THF (15 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (16 ml, 32 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C ab- gekühlt. Anschließend wurde eine Lösung aus Methyl-3-brompropanoat (5.41 g, 32.4 mmol) in THF (10 ml) langsam unter Rühren bei -78 °C hinzu getropft. Das Gemisch wurde über Nacht weiter gerührt, wobei das Kältebad (Trockeneis/Aceton) langsam auf RT kommen gelassen wurde. Anschließend wurde das Gemisch bei ca. 0 °C langsam mit Wasser und Ethylacetat (jeweils 100 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethyl- acetat (100 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung (150 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (50 g Kieselgel Biotage Snap-Cartridge Ultra, Cyclohexan/Ethylacetat-Gradient 93:7— > 7:3, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rück- stand im Vakuum getrocknet. Es wurden 4.97 g (98% Reinheit, 66% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.82 min
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 7.92-7.75 (m, 3H), 7.67-7.56 (m, 1 H), 4.39 (dd, 1 H), 3.59 (s, 3H), 2.57-2.43 (m, 2H, teilweise verdeckt), 2.37-2.23 (m, 1 H), 2.22-2.09 (m, 1 H). Beispiel 66A
(+/-)-ie/f-Butyl-4-cyan-4-(5-fluor-2-methylphenyl)butanoat (Racemat)
Zu einer Lösung aus (5-Fluor-2-methylphenyl)acetonitril (4.00 g, 26.8 mmol) in THF (30 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (16 ml, 32 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf - 78 °C abgekühlt. Anschließend wurde eine Lösung aus ie f-Butyl-3-brompropanoat (5.1 ml, 32
mmol) in THF (20 ml) langsam unter Rühren bei -78 °C hinzu getropft. Das Gemisch wurde über Nacht weiter gerührt, wobei das Kältebad (Trockeneis/Aceton) langsam auf RT kommen gelassen wurde. Anschließend wurde das Gemisch bei ca. 0 °C langsam mit Wasser und Ethylacetat (jeweils 100 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase ein- mal mit Ethylacetat (100 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung (150 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge Ultra, Cyclohe- xan/Ethylacetat-Gradient 93:7— > 7:3, Isolera One) gereinigt. Die vereinigten Zielfraktionen wur- den eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 4.94 g (100% Reinheit, 66% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.18 min; MS (ESIpos): m/z = 278 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 7.29 (dd, 1 H), 7.23 (dd, 1 H), 7.1 1 (td, 1 H), 4.34 (dd, 1 H), 2.42-2.34 (m, 2H), 2.30 (s, 3H), 2.17-1.96 (m, 2H), 1.40 (s, 9H). Beispiel 67A
(+/-)-ie/f-Butyl-4-cyan-4-[2-(trifluormethoxy)phenyl]butanoat (Racemat)
Zu einer Lösung aus [2-(Trifluormethoxy)phenyl]acetonitril (5.00 g, 24.9 mmol) in THF (65 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (15 ml, 30 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde eine Lösung aus ie f-Butyl-3-brompropanoat (4.7 ml, 30 mmol) in THF (45 ml) langsam unter Rühren bei -78 °C hinzu getropft. Das Gemisch wurde über Nacht weiter gerührt, wobei das Kältebad (Trockeneis/Aceton) langsam auf RT kommen gelassen wurde. Anschließend wurde das Gemisch bei ca. 0 °C langsam mit Wasser und Ethylacetat (jeweils 100 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (100 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung (150 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge Ultra, Cyclohe- xan/Ethylacetat-Gradient 93:7— > 7:3, Isolera One) gereinigt. Die vereinigten Zielfraktionen wur-
den eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 3.13 g (80% Reinheit, 31 % d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.27 min; MS (ESIpos): m/z = 330 [M+H]+ Beispiel 68A
(+/-)-ie/f-Butyl-4-cyan-4-(pyridin-2-yl)butanoat (Racemat)
Ansatz A: Pyridin-2-ylacetonitril (27 mg, 229 μηιοΙ), ie/f-Butyl-acrylat (50 μΙ, 340 μηιοΙ) und Kali- umcarbonat (63 mg, 457 μηηοΙ) wurden in DMF (0.3 ml) aufgenommen und 1 h in der Mikrowelle erhitzt (100 °C). Das Reaktionsgemisch wurde im Vakuum eingeengt. Das LC-MS Spektrum des Rohproduktes zeigte zu ca. 10% (UV) ein Produkt mit der gewünschten Masse.
Ansatz B: Pyrid i η-2-y laceton itri I (988 mg, 8.36 mmol) wurde in DMF (10 ml) aufgenommen und langsam mit Natriumhydrid (401 mg, 60% Reinheit, 10.0 mmol) versetzt (Gasentwicklung). Nach 10 min bei RT wurde ie f-Butyl-acrylat (1.2 ml, 8.4 mmol) langsam dazugegeben, und das Reaktionsgemisch wurde 3 h bei RT gerührt. Das Reaktionsgemisch wurde auf Wasser gegeben, mit Dichlormethan extrahiert, getrocknet (Natriumsulfat) und eingeengt. Das LC-MS Spektrum des Rohproduktes zeigte zu ca. 8% (UV) ein Produkt mit der gewünschten Masse.
Ansatz C: Im ausgeheizten Kolben wurde Tetrahydrofuran (10 ml) unter Stickstoff vorgelegt, Py- ridin-2-ylacetonitril (1.09 g, 9.23 mmol) dazugegeben und bei -78 °C langsam eine 2 M-Lösung von LDA in THF (6.9 ml, 14 mmol) zugetropft. Es wurde 10 min bei -78 °C und 20 min bei 0 °C nachgerührt und anschließend wieder auf -78 °C abgekühlt. Anschließend wurde sehr langsam ie f-Butyl-3-brompropanoat (2.32 g, 1 1.1 mmol) zugetropft und nach vollständiger Zugabe 1 h bei -78 °C nachgerührt. Das Kühlbad wurde entfernt, und das Reaktionsgemisch wurde 20 h weiter gerührt und dabei auf RT erwärmt. Das Gemisch wurde auf Wasser gegeben, mit Ethyl- acetat extrahiert, die organische Phase mit gesättigter Natriumchloridlösung gewaschen, ge- trocknet (Natriumsulfat) und eingeengt. Das LC-MS Spektrum des Rohproduktes zeigte zu ca. 17% (UV) ein Produkt mit der gewünschten Masse.
Die vereinigten Rohprodukte aus Ansatz A, Ansatz B und Ansatz C wurden chromatographisch aufgereinigt (Biotage Isolera Four, LiChroprep RP-18 (40-63 μηι); Gradient 5%— > 45% Aceto- nitril in 0.1 % wässriger Ammoniaklösung). Es wurden 799 mg (48% Reinheit, 9% d. Th.) der Ti- telverbindung erhalten.
LC-MS (Methode 6): Rt = 1.09 min; MS (ESIpos): m/z = 247 [M+H]+ Beispiel 69A
(+/-)-ie/f-Butyl-4-cyan-4-[2-fluor-6-(trifluormethyl)phenyl]butanoat (Racemat)

Die Reaktion wurde unter Argon durchgeführt. Zur Herstellung einer LDA-Lösung wurde eine Lösung von Diisopropylamin (6.2 ml, 44 mmol) in THF (27 ml) bei -15 °C langsam mit Butyllithi- um (Lösung 1.6 M in Hexan, 27 ml, 43 mmol) versetzt und das Gemisch 10 min bei 0 °C nachgerührt. Diese Lösung wurde langsam zu einer auf -78 °C gekühlten Lösung von [2-Fluor-6- (trifluormethyl)phenyl]acetonitril (8.01 g, 98% Reinheit, 38.7 mmol, CAS-RN 179946-34-0, kom- merziell verfügbar) in 74 ml THF hinzugetropft. Nach vollständiger Zugabe wurde das Kältebad entfernt, das Gemisch auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C gekühlt. Anschließend wurde langsam eine Lösung von ie f-Butyl-3-brompropanoat (8.0 ml, 97% Reinheit, 46 mmol) in 27 ml THF hinzugetropft und das Gemisch 1 h bei -78°C nachgerührt. Das Kältebad wurde entfernt und das Reaktionsgemisch über Nacht bei RT nachgerührt. Zur Aufarbei- tung wurde eine Ammoniumchlorid Lösung (10% in Wasser, 300 ml) hinzugefügt, das Gemisch 5 min kräftig verrührt und dann zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden nacheinander je zweimal mit 1 M Salzsäure, einer gesättigten Natriumhydrogen- carbonat-Lösung und einer gesättigten Natriumchlorid-Lösung gewaschen, dann über Natriumsulfat getrocknet und im Vakuum eingeengt. Der Rückstand wurde in einem Gemisch aus Cyclohexan, wenig Ethylacetat und Dichlormethan gelöst und mittels Flash-Chromatographie an Kieselgel gereinigt (Gradient Cyclohexan / Ethylacetat 100:0 bis 70:30). Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet und anschließend mittels präparativer HPLC (Methode 27) nachgereinigt. Die produkthaltigen Fraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 4.52 g (100% Reinheit, 35% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.23 min; MS (ESIpos): m/z = 332 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.219 (0.06), 1.379 (16.00), 1.536 (0.07), 2.073 (0.16), 2.091 (0.22), 2.107 (0.28), 2.125 (0.26), 2.144 (0.10), 2.213 (0.07), 2.231 (0.19), 2.250 (0.22), 2.267 (0.18), 2.285 (0.13), 2.302 (0.08), 2.327 (0.07), 2.365 (0.07), 2.384 (0.36), 2.393 (0.38), 2.402 (0.59), 2.410 (0.56), 2.420 (0.29), 2.428 (0.27), 2.452 (0.06), 2.669 (0.06), 4.373 (0.20), 4.390 (0.39), 4.407 (0.19), 7.698 (0.31 ), 7.71 1 (1.11 ), 7.722 (0.54), 7.748 (0.35), 7.766 (0.1 1 ).
Beispiel 70A
(+/-)-ie/f-Butyl-4-cyan-4-(2,3,5,6-tetrafluorphenyl)butanoat (Racemat)
Die Reaktion wurde unter Argon durchgeführt. Zur Herstellung einer LDA-Lösung wurde eine Lö- sung von Diisopropylamin (2.1 ml, 15 mmol) in THF (9 ml) bei -15 °C langsam mit Butyllithium (Lösung 1.6 M in Hexan, 9 ml, 14 mmol) versetzt und das Gemisch 10 min bei 0 °C nachgerührt. Diese Lösung wurde langsam zu einer auf -78 °C gekühlten Lösung von (2,3,5,6- Tetrafluorphenyl)acetonitril (2.47 g, 13.1 mmol, Beispiel 14A) in THF (25 ml) hinzugetropft. Nach vollständiger Zugabe wurde das Kältebad entfernt, das Gemisch auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C gekühlt. Anschließend wurde langsam eine Lösung von ie/f-Butyl- 3-brompropanoat (2.5 ml, 16 mmol) in THF (9 ml) hinzugetropft und das Gemisch 1 h bei -78 °C nachgerührt. Das Kältebad wurde entfernt und das Reaktionsgemisch über Nacht bei RT nachgerührt. Zur Aufarbeitung wurde eine Ammoniumchlorid-Lösung (10% in 100 ml Wasser) hinzugefügt, das Gemisch 5 min kräftig verrührt und dann zweimal mit Ethylacetat extrahiert. Die vereinig- ten organischen Phasen wurden nacheinander jeweils zweimal mit 1 M Salzsäure, einer gesättigten Natriumhydrogencarbonat-Lösung und einer gesättigten Natriumchlorid-Lösung gewaschen, dann über Natriumsulfat getrocknet und im Vakuum eingeengt. Der Rückstand wurde mittels Flash-Chromatographie an Kieselgel gereinigt (Isolera, 50 g Ultra Snap Säule, Cyclohexan / Ethylacetat Gradient). Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 3.43 g (100% Reinheit, 83% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.1 1 min; MS (ESIpos): m/z = 318 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.21 1 (0.06), 1.347 (1.57), 1.370 (16.00), 1.397 (0.08), 1.527 (0.06), 2.058 (0.05), 2.076 (0.15), 2.095 (0.19), 2.1 1 1 (0.29), 2.129 (0.28), 2.148 (0.1 1 ), 2.167 (0.08), 2.185 (0.27), 2.202 (0.33), 2.220 (0.25), 2.236 (0.15), 2.254 (0.08), 2.283 (0.02), 2.31 1 (0.06), 2.356 (0.68), 2.374 (1.21 ), 2.392 (0.47), 2.419 (0.03), 2.444 (0.04), 2.669 (0.03), 2.709 (0.03), 4.612 (0.25), 4.631 (0.50), 4.650 (0.24), 7.944 (0.06), 7.963 (0.13), 7.970 (0.14), 7.989 (0.27), 8.009 (0.15), 8.016 (0.15), 8.035 (0.08).
Beispiel 71A
(+/-)-ie/f-Butyl-4-cyan-4-(2,3,5-trifluorphenyl)butanoat (Racemat)
O CH3
Zu einer Lösung von (2,3,5-Trifluorphenyl)acetonitril (2.50 g, 97% Reinheit, 14.2 mmol, CAS-RN 243666-14-0, kommerziell verfügbar) in THF (15 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine LDA-Lösung (2 M in THF, 7.8 ml, 16 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde eine Lösung von ierf-Butyl-3-brompropanoat (3.36 g, 97% Reinheit, 15.6 mmol) in 5 ml THF langsam unter Rühren bei -78 °C hinzugetropft. Das Gemisch wurde über Nacht weiter gerührt, wobei das Kältebad (Trockeneis/Aceton) langsam auf RT kommen gelassen wurde. Anschließend wurde das Gemisch langsam mit Ethylacetat und Wasser (jeweils 100 ml) versetzt. Die Phasen wurde ge- trennt. Die wässrige Phase wurde zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet und eingeengt. Der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (Biotage 100 g Kieselgel-Kartusche, Cyclohexan / Ethylacetat Gradient 97:3 bis 90:10) gereinigt. Es wurden zwei Fraktionen erhalten, die getrennt im Vakuum ein- geengt wurden. Nach Trocknen im Vakuum wurden aus der zweiten Fraktion 600 mg (100% Reinheit, 14% d. Th.) einer ersten Charge der Titelverbindung erhalten. Die verunreinigte erste Fraktion wurde mittels präparativer HPLC (Methode 28) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurde 1.30 g (100% Reinheit, 31% d. Th., s. Analytik) einer zweiten Charge der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.15 min; MS (ESIpos): m/z = 300 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.220 (0.06), 1.379 (16.00), 1.536 (0.06), 2.051 (0.05), 2.070 (0.16), 2.088 (0.22), 2.104 (0.34), 2.122 (0.34), 2.141 (0.15), 2.165 (0.26), 2.182 (0.30), 2.200 (0.23), 2.216 (0.13), 2.235 (0.08), 2.329 (0.77), 2.348 (1.15), 2.366 (0.40), 2.671 (0.02), 2.71 1 (0.02), 4.489 (0.33), 4.508 (0.61 ), 4.526 (0.31 ), 7.253 (0.16), 7.259 (0.21 ), 7.267 (0.22), 7.272 (0.21 ), 7.281 (0.22), 7.287 (0.16), 7.293 (0.1 1 ), 7.576 (0.08), 7.583 (0.09), 7.592 (0.10), 7.599 (0.16), 7.603 (0.16), 7.612 (0.15), 7.620 (0.16), 7.625 (0.16), 7.632 (0.10), 7.639 (0.09), 7.647 (0.09).
Beispiel 72A
(+/-)-ie/f-Butyl-4-cyan-4-(2,3,6-trichlorphenyl)butanoat (Racemat)
Die Reaktion wurde unter Argon durchgeführt. Zur Herstellung einer LDA-Lösung wurde eine Lösung von Diisopropylamin (7.1 ml, 51 mmol) in THF (30 ml) bei -15 °C langsam mit Butyllithium (Lösung 1.6 M in Hexan, 30 ml, 48 mmol) versetzt, und das Gemisch wurde 10 min bei 0 °C nach- gerührt. Diese Lösung wurde langsam zu einer auf -78 °C gekühlten Lösung von (2,3,6- Trichlorphenyl)acetonitril (10.0 g, 97% Reinheit, 44.0 mmol, CAS-RN 3215-65-4, kommerziell verfügbar) in THF (84 ml) hinzugetropft. Nach vollständiger Zugabe wurde das Kältebad entfernt, das Gemisch auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C gekühlt. Anschließend wurde langsam eine Lösung von ierf-Butyl-3-brompropanoat (9.1 ml, 97% Reinheit, 53 mmol) in THF (30 ml) hinzugetropft und das Gemisch 1 h bei -78 °C nachgerührt. Das Kältebad wurde entfernt und das Reaktionsgemisch über Nacht bei RT nachgerührt. Zur Aufarbeitung wurde eine Ammoniumchlorid-Lösung (10% in 300 ml Wasser) hinzugefügt, das Gemisch 5 min kräftig verrührt und dann zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden nacheinander je zweimal mit 1 M Salzsäure, einer gesättigten Natriumhydrogencarbonat-Lösung und einer gesättigten Natriumchlorid-Lösung gewaschen und dann über Natriumsulfat getrocknet und im Vakuum eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 27) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 10.2 g (95% Reinheit, 63% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.41 min; MS (ESIpos): m/z = 348 [M+H]+
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: 1.382 (16.00), 1.997 (0.45), 2.316 (0.42), 2.367 (0.45), 2.381 (0.71 ), 2.403 (0.63), 3.322 (0.80), 5.002 (0.48), 7.620 (0.69), 7.637 (0.90), 7.746 (0.92), 7.763 (0.70).
Beispiel 73A
(+/-)-ie/f-Butyl-5-(2-chlorphenyl)-5-cyanpentanoat (Racemat)
Zu einer Lösung aus (2-Chlorphenyl)acetonitril (2.12 g, 14.0 mmol) in THF (20 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (8.4 ml, 17 mmol) ge-
geben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde eine Lösung aus ief-Butyl-4-brombutanoat (3.75 g, 16.8 mmol) in THF (10 ml) langsam unter Rühren bei -78 °C hinzu getropft. Das Gemisch wurde über Nacht weiter gerührt, wobei das Kältebad (Trockeneis/Aceton) langsam auf RT kommen gelassen wurde. An- schließend wurde das Gemisch bei ca. 0 °C langsam mit Wasser und Ethylacetat (jeweils 70 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (70 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung (150 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromato- graphie (100 g Kieselgel Biotage Snap-Cartridge Ultra, Cyclohexan/Ethylacetat-Gradient 93:7— > 7:3, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 2.70 g (96% Reinheit, 63% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.31 min; MS (ESIpos): m/z = 294 [M+H]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 7.62-7.50 (m, 2H), 7.49-7.37 (m, 2H), 4.49 (dd, 1 H), 2.28 (td, 2H), 1.99-1.78 (m, 2H), 1.72-1.51 (m, 2H), 1.38 (s, 9H).
Beispiel 74A
(+/-)-ie/f-Butyl-5-(2-chlorphenyl)-5-cyanhexanoat (Racemat)
Zu einer Lösung aus 2-(2-Chlorphenyl)propannitril (2.32 g, 14.0 mmol, CAS-RN 75920-46-6, kommerziell verfügbar) in THF (20 ml) unter Argon wurde bei -78 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (1 1 ml, 21 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C abgekühlt. Anschließend wurde eine Lösung aus ie f-Butyl-4-brombutanoat (3.75 g, 16.8 mmol) in THF (10 ml) langsam unter Rühren bei -78 °C hinzu getropft. Das Gemisch wurde über Nacht weiter gerührt, wobei das Kältebad (Trocken- eis/Aceton) langsam auf RT kommen gelassen wurde. Anschließend wurde das Gemisch bei ca. 0 °C langsam mit Wasser und Ethylacetat (jeweils 100 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (100 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung (150 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat-Gradient 93:7— > 7:3, Isolera One) gereinigt.
Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 2.47 g (98% Reinheit, 56% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.34 min; MS (ESIpos): m/z = 308 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.217 (0.06), 1.377 (16.00), 1.429 (0.19), 1.454 (0.1 1 ), 1.476 (0.30), 1.494 (0.46), 1.514 (0.33), 1.533 (0.21 ), 1.808 (3.89), 1.939 (0.15), 1.960 (0.23), 1.973 (0.21 ), 1.982 (0.16), 1.989 (0.18), 2.000 (0.20), 2.015 (0.16), 2.212 (0.19), 2.227 (0.84), 2.244 (1.47), 2.254 (0.27), 2.262 (0.65), 2.288 (0.13), 7.392 (0.13), 7.405 (0.36), 7.410 (0.45), 7.414 (0.39), 7.421 (0.91 ), 7.429 (0.53), 7.433 (0.48), 7.438 (0.49), 7.451 (0.17), 7.528 (0.54), 7.534 (0.41 ), 7.547 (0.27), 7.552 (0.40), 7.564 (0.52), 7.569 (0.34), 7.582 (0.35), 7.588 (0.37). Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 7.61-7.51 (m, 2H), 7.48-7.36 (m, 2H), 2.31-2.18 (m, 3H), 2.04-1.92 (m, 1 H), 1.81 (s, 3H), 1.57-1.44 (m, 2H), 1.38 (s, 9H).
Beispiel 75A
(+/-)-Methyl-3-amino-2-phenylpropanoat-Hydrochlorid (Racemat)
Zu einer Suspension aus (+/-)-3-Amino-2-phenylpropansäure (700 mg, 4.24 mmol) in Methanol (10 ml) wurde bei RT Oxalylchlorid (740 μΙ, 8.5 mmol) langsam hinzugetropft, und das Gemisch wurde 5 Tage bei RT gerührt. Anschließend wurde das Gemisch eingeengt. Der Rückstand wurde mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Anschließend wurde der Rückstand im Vakuum getrocknet. Es wurden 933 mg (95% Reinheit, 97% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 4): Rt = 0.93 min; MS (ESIpos): m/z = 180 [M+H]+
1 H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.20 (br. s, 3H), 7.63-7.10 (m, 5H), 4.1 1 (dd, 1 H), 3.64 (s, 3H), 3.46 (dd, 1 H), 3.07 (dd, 1 H).
Beispiel 76A
(+/-)-Ethyl-3-amino-2-(2-chlorphenyl)propanoat (Racemat)
Eine Lösung aus (+/-)-Ethyl-(2-chlorphenyl)(cyan)acetat (8.60 g, 38.5 mmol, Beispiel 15A) in Ethanol (140 ml) wurde mit Raney-Nickel (1.13 g, 19.2 mmol) versetzt und 20 h bei 3 bar hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert, mit Ethanol gewaschen und die Mutter- lauge eingeengt. Der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (200 g Kieselgel Biotage Snap-Cartridge KP-Sil, Dichlormethan/Methanol 40:1 ) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 869 mg (87% Reinheit, 9% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 4): Rt = 1.19 min; MS (ESIpos): m/z = 228 [M+H]+
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 7.50-7.44 (m, 1 H), 7.43-7.26 (m, 3H), 4.15-4.03 (m, 3H), 3.10 (dd, 1 H), 2.83 (dd, 1 H), 1.54 (br. s, 2H), 1.14 (t, 3H).
Beispiel 77A
(+/-)-Methyl-3-amino-2-(2-methoxyphenyl)propanoat-Hydrochlorid (Racemat)
Zu einer Lösung aus (+/-)-3-Amino-2-(2-methoxyphenyl)propansäure-Hydrochlorid (1.39 g, 6.00 mmol, CAS-RN 91012-74-7, kommerziell verfügbar) in Methanol (14 ml, 350 mmol) wurde unter Kühlung mit einem Eis-Aceton-Bad langsam Oxalylchlorid (1.0 ml, 12 mmol) hinzugetropft, und das Gemisch wurde 24 h bei RT gerührt. Anschließend wurde das Gemisch eingeengt. Der Rückstand wurde mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Anschließend wurde der Rückstand im Vakuum getrocknet. Es wurden 1 .83 g (89% Reinheit, „>100% d. Th.", lösungsmittelhaltig) der Titelverbindung erhalten.
LC-MS (Methode 4): Rt = 0.94 min; MS (ESIpos): m/z = 210 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 3.373 (1.52), 3.394 (1.49), 3.425 (1.14), 3.716 (5.40), 3.766 (1.44), 3.780 (16.00), 3.805 (5.37), 6.936 (0.77), 6.939 (0.84), 6.955 (1.68), 6.958 (1.78),
6.974 (0.96), 6.976 (0.99), 7.042 (1.55), 7.063 (1.84), 7.182 (1.39), 7.187 (1.54), 7.201 (1.21 ), 7.205 (1.25), 7.306 (0.96), 7.31 1 (0.89), 7.325 (1.25), 7.345 (0.73), 7.350 (0.64), 8.079 (0.97).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.08 (br. s, 2H), 7.36-7.29 (m, 1 H), 7.19 (dd, 1 H), 7.05 (d, 1 H), 6.99-6.92 (m, 1 H), 4.30-4.22 (m, 1 H), 3.81 (s, 1 H), 3.78 (s, 3H), 3.72 (s, 1 H), 3.62 (s, 3H), 3.45-3.35 (m, 1 H, verdeckt), 3.02-2.93 (m, 1 H).
Beispiel 78A
(+/-)-Methyl-3-amino-2-(4-fluorphenyl)propanoat-Hydrochlorid (Racemat)
Zu einer Lösung aus (+/-)-3-Amino-2-(4-fluorphenyl)propansäure-Hydrochlorid (500 mg, 2.28 mmol, CAS-Nr. 15032-53-8) in Methanol (5.4 ml, 130 mmol) wurde unter Kühlung mit einem Eis-Aceton-Bad langsam Oxalyichlorid (400 μΙ, 4.6 mmol) hinzugetropft, und das Gemisch wurde 24 h bei RT gerührt. Anschließend wurde das Gemisch eingeengt. Der Rückstand wurde mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Anschließend wurde der Rückstand im Vakuum getrocknet. Es wurden 570 mg (100% Reinheit,„>100% d. Th.", lö- sungsmittelhaltig) der Titelverbindung erhalten.
LC-MS (Methode 4): Rt = 0.99 min; MS (ESIpos): m/z = 198 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 3.428 (0.95), 3.449 (0.95), 3.639 (16.00), 3.680 (1.34), 7.204 (1.44), 7.226 (3.36), 7.248 (1.99), 7.342 (2.08), 7.348 (0.90), 7.355 (2.25), 7.364 (1.75), 7.377 (1.52), 8.125 (1.09). Beispiel 79A
(+/-)-Methyl-3-amino-2-hydroxy-2-phenylpropanoat-Hydrochlorid (Racemat)
Durch eine Suspension aus (+/-)-3-Amino-2-hydroxy-2-phenylpropansäure (4.0 g, 22.10 mmol, darstellbar gemäß Justus Liebigs Annalen der Chemie 1961 , 639, 166-180) in Methanol (600 ml) wurde 15 min Chlorwasserstoff geleitet. Dabei ging die Suspension in eine Lösung über. Anschließend wurde das Reaktionsgemisch 2.5 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das Gemisch eingeengt, und der Rückstand wurde in ie f-Butylmethylether verrührt. Der vorhandene Feststoff wurde abfiltriert und im Vakuum getrocknet. Es wurden 2.81 g (55% d. Th.) der Titelverbindung erhalten.
Beispiel 80A
(+/-)-Ethyl-3-amino-2-[3-(trifluormethyl)phenyl]propanoat (Racemat)
Eine Lösung aus (+/-)-Ethyl-cyan[3-(trifluormethyl)phenyl]acetat (1 .26 g, 4.90 mmol, darstellbar gemäß US4909830, 1990 und European Journal of Organic Chemistry 2014, 27, 6025-6029) in ie/f-Butanol (20 ml) wurde mit Raney-Nickel (288 mg, 4.90 mmol) versetzt und zwei Tage bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert, die Mutter- lauge eingeengt und der Rückstand im Vakuum getrocknet. Der Rückstand wurde mittels präpa- rativer HPLC (Methode 22) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 63 mg (100% Reinheit, 5% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 4): Rt = 1 .43 min; MS (ESIpos): m/z = 262 [M+H]+
Beispiel 81A
(+/-)-Ethyl-3-amino-2-(1 ,3-benzodioxol-5-yl)propanoat (Racema,
Eine Lösung aus (+/-)-Ethyl-1 ,3-benzodioxol-5-yl(cyan)acetat (750 mg, 3.22 mmol, darstellbar gemäß Beilstein Journal of Organic Chemistry 2015, 1 1 , 875-883) in ie f-Butanol (13 ml) wurde mit Raney-Nickel (189 mg, 3.22 mmol) versetzt und zwei Tage bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert, die Mutterlauge eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 150 mg (100% Reinheit, 20% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 0.71 min; MS (ESIpos): m/z = 238 [M+H]+ Beispiel 82A
(+/-)-ie/f-Butyl-3-amino-2-(3-chlorpyridin-2-yl)-2-methylpropanoat (Racemat)
Eine Lösung aus (+/-)-fe/f-Butyl-2-(3-chlorpyridin-2-yl)-2-cyanpropanoat (900 mg, nicht reinheits- korrigiert,„3.37 mmol", Beispiel 16A) in ie f-Butanol (20 ml) wurde mit Raney-Nickel (198 mg, 3.37 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert und die Mutterlauge eingeengt. Der Rückstand wurde im Vakuum getrocknet und anschließend mittels präparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 255 mg (100% Reinheit, 28% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.04 min; MS (ESIpos): m/z = 271 [M+H]+ Beispiel 83A
(+/-)-ie/f-Butyl-3-amino-2-(2,6-difluorphenyl)-2-methylpropanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-2-cyan-2-(2,6-difluorphenyl)propanoat (6.80 g, 79% Reinheit, 20.1 mmol, Beispiel 17A) in ie/f-Butanol (22 ml) wurde mit Raney-Nickel (1.03 g, 17.5 mmol) versetzt und drei Tage bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert und die Mutterlauge eingeengt. Der Rückstand wurde in Ethylacetat aufgenom-
men und zweimal mit 1 M Salzsäure extrahiert. Die vereinten wässrigen Phasen wurden mit 20%-iger Natronlauge alkalisch eingestellt und danach zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden über Magnesiumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 1.40 g (99% Reinheit, 25% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 0.57 min; MS (ESIpos): m/z = 216 [M-'Bu+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.368 (16.00), 1.497 (1.28), 1.502 (2.25), 1.508 (1.24), 2.990 (0.85), 2.998 (0.74), 7.005 (0.55), 7.026 (0.72), 7.031 (0.61 ), 7.052 (0.67), 7.332 (0.44).
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 7.39-7.28 (m, 1 H), 7.08-6.98 (m, 2H), 3.05-2.94 (m, 2H), 1.50 (t, 3H), 1.37 (s, 9H).
Beispiel 84A
(+/-)-Ethyl-3-amino-2-methyl-2-[3-(trifluormethyl)phenyl]propanoat (Racemat)
Eine Lösung aus (+/-)-Ethyl-2-cyan-2-[3-(trifluormethyl)phenyl]propanoat (1.80 g, 6.64 mmol, darstellbar gemäß WO 2016/94260 A1 , S. 82, in Analogie zur Darstellung von (+/-)-Ethyl-2-cyan- 2-(4-fluorophenyl)propanoat) in ie/f-Butanol (39 ml) wurde mit Raney-Nickel (390 mg, 6.64 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert, die Mutterlauge eingeengt und der Rückstand im Vakuum getrocknet. Der Rückstand wurde mittels präparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfrak- tionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 615 mg (96% Reinheit, 32% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 0.55 min; MS (ESIpos): m/z = 276 [M+H]+
Beispiel 85A
(+/-)-ie/f-Butyl-3-amino-2-(3-methoxyphenyl)propanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-cyan(3-methoxyphenyl)acetat (4.00 g, 16.2 mmol, Beispiel 18A) in ie f-Butanol (75 ml) wurde mit Raney-Nickel (1.00 g, 17.03 mmol) versetzt und 5 Tage bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert, die Mutter- lauge eingeengt und der Rückstand im Vakuum getrocknet. Der Rückstand wurde danach in Ethylacetat aufgenommen und mit 1 M Salzsäure extrahiert. Anschließend wurde die wässrige Phase mit konzentrierter Natronlauge alkalisch gestellt und mit Ethylacetat extrahiert. Die vereinten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 750 mg (89% Reinheit, 16% d. Th.) der Ti- telverbindung erhalten.
LC-MS (Methode 2): Rt = 0.59 min; MS (ESIpos): m/z = 252 [M+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 7.24 (t, 1 H), 6.86-6.76 (m, 3H), 3.73 (s, 3H), 3.48 (dd, 1 H), 3.01 (dd, 1 H), 2.73 (dd, 1 H), 1.37 (s, 9H).
Beispiel 86A
(+/-)-ie/f-Butyl-3-amino-2-(3-chlorphenyl)propanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-(3-chlorphenyl)(cyan)acetat (5.50 g, „21.9 mmol", nicht rein- heitskorrigiert, Beispiel 19A) in ie f-Butanol (95 ml) wurde mit Raney-Nickel (1.28 g, 21.9 mmol) versetzt und 5 Tage bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kiesel- gur abfiltriert, die Mutterlauge eingeengt und der Rückstand im Vakuum getrocknet. Der Rückstand wurde danach in Ethylacetat (50 ml) aufgenommen und mit 1 M Salzsäure (80 ml) extrahiert. Anschließend wurde die wässrige Phase mit konzentrierter Natronlauge alkalisch gestellt und mit Ethylacetat (70 ml) extrahiert. Die vereinten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 500 mg (95% Reinheit,„9%" d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.1 1 min; MS (ESIpos): m/z = 200 [M-'Bu+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 7.42-7.29 (m, 3H), 7.26-7.20 (m, 1 H), 3.60 (t, 1 H), 3.08 (dd, 1 H), 2.81 (dd, 1 H), 1.37 (s, 9H).
Beispiel 87A
(+/-)-ie/f-Butyl-3-amino-2-(3-methylphenyl)propanoat (Racemat)
Eine Lösung aus (+/-)-ierf-Butyl-cyan(3-methylphenyl)acetat (3.20 g, 13.8 mmol, Beispiel 20A) in ie/f-Butanol (60 ml) wurde mit Raney-Nickel (812 mg, 13.8 mmol) versetzt und 5 Tage bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert, die Mutterlauge eingeengt und der Rückstand im Vakuum getrocknet. Der Rückstand wurde danach in Ethyl- acetat (50 ml) aufgenommen und mit 1 M Salzsäure (80 ml) extrahiert. Anschließend wurde die wässrige Phase mit konzentrierter Natronlauge alkalisch gestellt und mit Ethylacetat (70 ml) extrahiert. Die vereinten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 700 mg (90% Reinheit, 19% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 0.63 min; MS (ESIpos): m/z = 236 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.366 (16.00), 1.384 (0.46), 1.399 (0.45), 2.282 (4.17), 3.451 (0.41 ), 7.019 (0.47), 7.049 (1.41 ), 7.068 (0.54), 7.185 (0.44), 7.203 (0.64).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 7.25-7.15 (m, 1 H), 7.09-6.99 (m, 3H), 3.45 (dd, 1 H), 3.02 (dd, 1 H), 2.71 (dd, 1 H), 2.28 (s, 3H), 1.37 (s, 9H). Beispiel 88A
(+/-)-ie/f-Butyl-3-amino-2-(2-fluor-6-methoxyphenyl)propanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-cyan(2-fluor-6-methoxyphenyl)acetat (1.04 g, 62% Reinheit, 2.43 mmol, Beispiel 21 A) in Essigsäure (10 ml) wurde mit Palladium auf Kohle (26 mg, 10% Reinheit, 24.3 μηηοΙ) versetzt und 24 h bei Normaldruck hydriert. Anschließend wurde der Kata-
lysator über Kieselgur abfiltriert und zweimal mit Essigsäure (jeweils 2 ml) gewaschen. Das Filt- rat wurde mit Wasser (100 ml) verdünnt und zweimal mit Ethylacetat (jeweils 50 ml) extrahiert. Anschließend wurde die wässrige Phase mit Natronlauge auf pH 8 eingestellt und zweimal mit Ethylacetat (jeweils 100 ml) extrahiert. Die vereinigten organischen Phasen wurden über Natri- umsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 222 mg (71 % Reinheit, 24% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 0.96 min; MS (ESIpos): m/z = 270 [M+H]+ Beispiel 89A
(+/-)-ie/f-Butyl-3-amino-2-(2-methoxyphenyl)-2-methylpropanoat (Racemat)
Eine Lösung aus (+/-)-ierf-Butyl-2-cyan-2-(2-methoxyphenyl)propanoat (1.30 g, 4.97 mmol, Beispiel 22A) in ie f-Butanol (25 ml) wurde mit Raney-Nickel (292 mg, 4.97 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert und die Mutterlauge eingeengt. Der Rückstand wurde im Vakuum getrocknet. Es wurden 1.31 g (70% Reinheit, 69% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.02 min; MS (ESIpos): m/z = 266 [M+H]+ Beispiel 90A
(+/-)-ie/f-Butyl-3-amino-2-(2-chlor-6-fluorphenyl)-2-methylpropanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-2-(2-chlor-6-fluorphenyl)-2-cyanpropanoat (1.30 g, 4.58 mmol, Beispiel 23A) in ie f-Butanol (26 ml) wurde mit Raney-Nickel (269 mg, 4.58 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert und die Mutterlauge eingeengt. Der Rückstand wurde im Vakuum getrocknet. Es wurden 1.25 g (70% Reinheit, 67% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.08 min; MS (ESIpos): m/z = 288 [M+H]+
Beispiel 91A
(+/-)-fe/f-Butyl-4-amino-3-(2-chlorphenyl)butanoat (Racemat)
Eine Lösung aus (+/-)-ferf-Butyl-3-(2-chlorphenyl)-3-cyanpropanoat (100 mg, 376 μηιοΙ, Beispiel 24A) in ie f-Butanol (10 ml) wurde mit Raney-Nickel (22 mg, 376 μηηοΙ) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert und die Mutterlauge mittels praparativer HPLC (Methode 22) gereinigt. Es wurden 87 mg (Reinheit 100%, 86% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 0.99 min; MS (ESIpos): m/z = 270 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.200 (16.00), 1.212 (0.81 ), 2.683 (0.47), 2.708 (0.49), 2.81 1 (0.49), 2.825 (0.51 ), 3.053 (0.47), 7.305 (0.48), 7.308 (0.50), 7.365 (0.52), 7.460 (1.19), 7.480 (0.88), 7.483 (0.77), 7.939 (0.44).
Beispiel 92A
(+/-)-ie/f-Butyl-4-amino-3-(3-chlorphenyl)butanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-3-(3-chlorphenyl)-3-cyanpropanoat (710 mg, 2.67 mmol, Beispiel 25A) in ie f-Butanol (1 1.6 ml) wurde mit Raney-Nickel (157 mg, 2.67 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert und die Mutterlauge eingeengt. Der Rückstand wurde im Vakuum getrocknet. Es wurden 294 mg (Reinheit 40%, 16% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.39 min; MS (ESIpos): m/z = 270 [M+H]+ Beispiel 93A
(+/-)-ie/f-Butyl-4-amino-3-(2-methylphenyl)butanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-3-cyan-3-(2-methylphenyl)propanoat (850 mg, 3.46 mmol, Beispiel 26A) in ie f-Butanol (15 ml) wurde mit Raney-Nickel (203 mg, 3.46 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert und die Mutterlauge eingeengt. Es wurden 724 mg (95% Reinheit, 80% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 4): Rt = 1.32 min; MS (ESIpos): m/z = 250 [M+H]+
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 7.18-7.02 (m, 4H), 3.31-3.22 (m, 1 H), 2.76 (dd, 1 H), 2.70-2.55 (m, 2H), 2.40 (dd, 1 H), 2.32 (s, 3H), 1.18 (s, 9H) Beispiel 94A
(+/-)-ie/f-Butyl-4-amino-3-(2,6-dichlorphenyl)butanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-3-cyan-3-(2,6-dichlorphenyl)propanoat (680 mg, 2.27 mmol, Beispiel 27A) in ie f-Butanol (10 ml) wurde mit Raney-Nickel (133 mg, 2.27 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert und die Mutterlauge eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 25) gereinigt. Es wurden 255 mg (94% Reinheit, 35% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 4): Rt = 1.46 min; MS (ESIpos): m/z = 304 [M+H]+
Beispiel 95A
(+/-)-ie/f-Butyl-4-amino-3-(3-methoxyphenyl)butanoat (Racemat)
Eine Lösung aus (+/-)-fe/f-Butyl-3-cyan-3-(3-methoxyphenyl)propanoat (389 mg, 1.49 mmol, Beispiel 28A) in fe/f-Butanol (6.5 ml) wurde mit Raney-Nickel (88 mg, 1.49 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert und die Mutterlauge eingeengt und im Vakuum getrocknet. Es wurden 500 mg („90% Rein- heit",„>100% d. Th.", lösungsmittelhaltig) der Titelverbindung erhalten.
LC-MS (Methode 4): Rt = 1.26 min; MS (ESIpos): m/z = 266 [M+H]+ Beispiel 96A
(+/-)-ie/f-Butyl-4-amino-3-(3-chlorpyridin-2-yl)-3-methylbutanoat (Racemat)
Eine Lösung aus (+/-)-fe/f-Butyl-3-(3-chlorpyridin-2-yl)-3-cyanbutanoat (1.00 g, 3.56 mmol, Beispiel 29A) in ie f-Butanol (21 ml) wurde mit Raney-Nickel (209 mg, 3.56 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert und die Mutterlauge mittels präparativer HPLC (Methode 16) gereinigt. Es wurden 616 mg (100% Reinheit, 61 % d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 0.62 min; MS (ESIpos): m/z = 285 [M+H]+ Beispiel 97A
(+/-)-ie/f-Butyl-4-amino-3-(2-chlor-6-fluorphenyl)butanoat (Racemat)
Methode A:
Eine Lösung aus (+/-)-fe/f-Butyl-3-(2-chlor-6-fluorphenyl)-3-cyanpropanoat (870 mg, 3.07 mmol, Beispiel 30A) in ie f-Butanol (13.4 ml) wurde mit Raney-Nickel (180 mg, 3.07 mmol) versetzt und drei Tage bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert und die Mutterlauge eingeengt. Es wurden 551 mg (30% Reinheit, 19% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.02 min; MS (ESIpos): m/z = 288 [M+H]+
Methode B:
Eine Lösung aus (+/-)-fe/f-Butyl-3-(2-chlor-6-fluorphenyl)-3-cyanpropanoat (43.9 g, 79% Reinheit, 122 mmol, Beispiel 30A) in ie/f-Butanol (500 ml) wurde mit Raney-Nickel (7.15 g, 122 mmol) versetzt und vier Tage bei Normaldruck hydriert. Anschließend wurde erneut Raney- Nickel (7.15 g, 122 mmol) hinzugegeben und das Gemisch wurde einen weiteren Tag bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert und zweimal mit ie f-Butanol (15 ml) gewaschen. Das Filtrat wurde eingeengt, und der Rückstand wurde in Ethylacetat (300 ml) aufgenommen und zweimal mit 1 M Salzsäure (250 ml) extrahiert. Die vereinigten wässigen Phasen wurden mit gesättigter Natriumcarbonat-Lösung auf pH 8-9 eingestellt und zweimal mit Ethylacetat (200 ml) extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt. Es wurden 20.1 1 g (98% Reinheit, 57% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 0.55 min; MS (ESIpos): m/z = 288 [M+H]+
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 7.36-7.22 (m, 2H), 7.21-7.07 (m, 1 H), 3.73-3.54 (m, 1 H), 2.87-2.72 (m, 3H), 2.70-2.56 (m, 1 H), 1.20 (s, 9H).
Beispiel 98A
(+/-)-ie/f-Butyl-4-amino-3-(4-methylphenyl)butanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-3-cyan-3-(4-methylphenyl)propanoat (280 mg, 90% Reinheit, 1.03 mmol, Beispiel 31 A) in ie f-Butanol (4.5 ml) wurde mit Raney-Nickel (60 mg, 1.03 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert und die Mutterlauge eingeengt. Es wurden 106 mg (97% Reinheit, 40% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 0.66 min; MS (ESIpos): m/z = 250 [M+H]+ Beispiel 99A
(+/-)-ie/f-Butyl-4-amino-3-(3-methylphenyl)butanoat (Racemat)
Eine Lösung aus (+/-)-fe/f-Butyl-3-cyan-3-(3-methylphenyl)propanoat (174 mg, 709 μηηοΙ, Beispiel 32A) in ie f-Butanol (3.1 ml) wurde mit Raney-Nickel (42 mg, 709 μηηοΙ) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert und die Mutterlauge eingeengt. Es wurden 171 mg (77% Reinheit, 74% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.02 min; MS (ESIpos): m/z = 250 [M+H]+ Beispiel 100A
(+/-)-fe/f-Butyl-4-amino-3-(2-methoxyphenyl)butanoat (Racemat)
Eine Lösung aus (+/-)-fe/f-Butyl-3-cyan-3-(2-methoxyphenyl)propanoat (4.80 g, 18.4 mmol, Beispiel 33A) in ie f-Butanol (80 ml) wurde mit Raney-Nickel (1.08 g, 18.4 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert und die Mutterlauge eingeengt. Es wurden 4.23 g (92% Reinheit, 80% d. Th.) der Titelverbin- dung erhalten.
LC-MS (Methode 2): Rt = 0.57 min; MS (ESIpos): m/z = 266 [M+H]+
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 7.21-7.14 (m, 1 H), 7.11 (dd, 1 H), 6.94 (d, 1 H), 6.87 (td, 1 H), 3.76 (s, 3H), 3.45-3.33 (m, 1 H), 2.75-2.65 (m, 2H), 2.61 (dd, 1 H), 2.43 (dd, 1 H), 1.22 (s, 9H).
Beispiel 101A
(+/-)-ie/f-Butyl-4-amino-3-[2-(trifluormethoxy)phenyl]butanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-3-cyan-3-[2-(trifluormethoxy)phenyl]propanoat (6.70 g, 65% Reinheit, 13.8 mmol, Beispiel 34A) in ie/f-Butanol (14 ml) wurde mit Raney-Nickel (669 mg, 11.4 mmol) versetzt und 24 h bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert und die Mutterlauge eingeengt. Der Rückstand wurde in Ethylacetat aufge- nommen und zweimal mit 1 M Salzsäure extrahiert. Die vereinten wässrigen Phasen wurden mit 20%-iger Natronlauge alkalisch eingestellt und danach zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden über Magnesiumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 700 mg (100% Reinheit, 16% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 0.71 min; MS (ESIpos): m/z = 320 [M+H]+
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 7.46-7.39 (m, 1 H), 7.38-7.26 (m, 3H), 3.43-3.33 (m, 1 H), 2.80 (dd, 1 H), 2.75-2.59 (m, 2H), 2.42 (dd, 1 H), 1.22 (s, 9H).
Beispiel 102A
(+/-)-ie/f-Butyl-4-amino-3-(pyridin-2-yl)butanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-3-cyan-3-(pyridin-2-yl)propanoat (1.90 g, 8.18 mmol, Beispiel 35A) in ief-Butanol (35 ml) wurde mit Raney-Nickel (480 mg, 8.18 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert und die Mutterlauge eingeengt. Der Rückstand wurde mittels präparativer HPLC [Säule: XBridge C18, 5 μηη, 100 mm x 30 mm; Fluss: 75 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 0.60 ml; Eluent: 90% Wasser / 5% (Aceton itril/Wasser 8:2) + 1% Ammoniak-Lösung / 5% Acetonitril -> 0% Wasser / 5% (Acetonitril/Wasser 8:2) + 1 % Ammoniak-Lösung / 95% Acetonitril; Laufzeit 8.5 min] gereinigt. Es wurden 559 mg einer ersten Charge der Titelverbindung (80% Reinheit, 23% d. Th., siehe LC- MS) und 383 mg einer zweiten Charge der Titelverbindung (27% Reinheit, 5% d. Th.) erhalten. LC-MS (Methode 4): Rt = 1.06 min; MS (ESIpos): m/z = 237 [M+H]+
Beispiel 103A
(+/-)-ie/f-Butyl-4-amino-3-[2-(trifluormethyl)phenyl]butanoat (Racemat)
Eine Lösung aus (+/-)-ierf-Butyl-3-cyan-3-[2-(trifluormethyl)phenyl]propanoat (2.00 g, 6.68 mmol, Beispiel 36A) in ie f-Butanol (39 ml) wurde mit Raney-Nickel (392 mg, 6.68 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde erneut Raney-Nickel (392 mg, 6.68 mmol) hinzugegeben, und das Gemisch wurden weitere 24 h bei Normaldruck hydriert. Danach wurde der Katalysator über Kieselgur abfiltriert, das Filtrat eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 1.90 g (Reinheit 60%, 56% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.16 min; MS (ESIpos): m/z = 304 [M+H]+ Beispiel 104A
(+/-)-ie/f-Butyl-4-amino-3-(3-methoxypyridin-2-yl)butanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-3-cyan-3-(3-methoxypyridin-2-yl)propanoat (2.20 g, 8.39 mmol, Beispiel 37A) in ie f-Butanol (37 ml) wurde mit Raney-Nickel (492 mg, 8.39 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfil- triert und die Mutterlauge eingeengt und im Vakuum getrocknet. Es wurden 2.02 g (95% Reinheit, 86% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 4): Rt = 1.12 min; MS (ESIpos): m/z = 267 [M+H]+ Beispiel 105A
(+/-)-ie/f-Butyl-3-(2-chlorphenyl)-2,2-dimethyl-4-nitrobutanoat (Racemat)
Zu einer 2 M Lösung aus LDA in THF/Heptane/Ethylbenzene unter Argon wurde bei -78 °C eine Lösung aus ie f-Butyl-2-methylpropanoat (51 1 mg, 3.54 mmol) in THF (2 ml) langsam hinzugetropft, und das Gemisch wurde 30 min bei -78 °C gerührt. Anschließend wurde eine Lösung aus 1-Chlor-2-(2-nitrovinyl)benzol (500 mg, 2.72 mmol, CAS-Nummer 3156-34-1 ) in THF (2 ml) hin- zugetropft, und das Gemisch wurde weitere 30 min bei -78 °C gerührt. Anschließend wurde das Gemisch mit Eisessig und dann mit Wasser versetzt, auf RT kommen gelassen und dann mehrmals mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde mittels Flash-Säulenchromatographie (50 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/ Ethylacetat 80:20) gereinigt. Es wurden 590 mg (88% Reinheit, 58% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 7): Rt = 1.61 min; MS (ESIneg): m/z = 326 [M-H]-
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 7.54 (dd, 1 H), 7.47 (dd, 1 H), 7.40-7.28 (m, 2H), 5.14 (dd, 1 H), 4.91 (dd, 1 H), 4.49 (dd, 1 H), 1.42 (s, 9H), 1.08 (s, 3H), 1.06 (s, 3H).
Beispiel 106A
(+/-)-ie/f-Butyl-4-amino-3-(2-chlorphenyl)-2,2-dimethylbutanoat (Racen
Eine Lösung aus (+/-)-ierf-Butyl-3-(2-chlorphenyl)-2,2-dimethyl-4-nitrobutanoat (595 mg, 1.82 mmol, Beispiel 105A) in Methanol (10 ml) wurde mit Raney-Nickel (geschätzt 200 mg, 3.4 mmol) versetzt und 3 h bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert, mit Methanol gewaschen und die Mutterlauge mittels präparativer HPLC (Methode: Chromatorex C18, 10 μηι, 30 x 125 mm, Acetonitril / (Wasser + 0.005% Ammoniak- Gradient: Acetonitril 10% -> 90%) gereinigt. Es wurden 320 mg (Reinheit 100%, 86% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 8): Rt = 5.00 min; MS (ESIpos): m/z = 298 [M+H]+
LC-MS (Methode 7): Rt = 0.86 min; MS (ESIpos): m/z = 242 [M-'Bu+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 7.45 (dd, 1 H), 7.39-7.30 (m, 2H), 7.28-7.22 (m, 1 H), 3.72 (dd, 1 H), 2.98-2.89 (m, 1 H), 2.85-2.79 (m, 1 H), 1.40 (s, 9H), 1.01 (s, 3H), 0.97 (s, 3H).
Beispiel 107A
(+/-)-ie/f-Butyl-4-amino-3-(2-chlorphenyl)-3-methylbutanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-3-(2-chlorphenyl)-3-cyanbutanoat (5.00 g, 17.9 mmol, Beispiel 38A) in ie f-Butanol (100 ml) wurde mit Raney-Nickel (1.05 g, 17.9 mmol) versetzt und 24 h bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert, die Mutterlauge eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 3.58 g (70% Reinheit, 49% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.05 min; MS (ESIpos): m/z = 284 [M+H]+
Beispiel 108A
(+/-)-ie/f-Butyl-4-amino-3-(2-fluorphenyl)-3-methylbutanoat (Racen
Eine Lösung aus (+/-)-ie/f-Butyl-3-(2-chlor-6-fluorphenyl)-3-cyanbutanoat (4.00 g, 13.4 mmol, Beispiel 39A) in ie f-Butanol (76 ml) wurde mit Raney-Nickel (789 mg, 13.4 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert, die Mutterlauge eingeengt und der Rückstand mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat 85:15) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 2.51 g (75% Reinheit, 51 % d. Th.) der Titelverbindung im Gemisch mit ie/f-Butyl-4-amino-3-(2-chlor- 6-fluorphenyl)-3-methylbutanoat (zu 10% laut LC-MS enthalten) erhalten.
LC-MS (Methode 1 ): Rt = 0.90 min; MS (ESIpos): m/z = 268 [M+H]+
Beispiel 109A
(+/-)-ie/f-Butyl-4-amino-3-methyl-3-(pyridin-2-yl)butanoat (Racemat)
Eine Lösung aus (+/-)-fe/f-Butyl-3-(3-chlorpyridin-2-yl)-3-cyanbutanoat (14.8 g, 52.7 mmol), Beispiel 29A) in ief-Butanol (300 ml) wurde mit Raney-Nickel (3.09 g, 52.7 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert, die Mutterlauge eingeengt und der Rückstand mittels präparativer HPLC (Methode 16) gereinigt. Es wurden 5.55 g einer Fraktion isoliert, in der überwiegend unreagiertes Edukt (+/-)-fe/f-Butyl-3-(3- chlorpyridin-2-yl)-3-cyanbutanoat enthalten war. Diese Fraktion wurde erneut in fe/f-Butanol (200 ml) gelöst, mit Raney-Nickel (3.09 g, 52.7 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert. Die Mutterlauge wurde eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 4.0 g der Titelverbindung (11 % Reinheit, ca. 3% d. Th.) im Gemisch mit ie/f-Butyl-4-amino-3-(3-chlorpyridin-2-yl)-3-methylbutanoat (zu 75% laut LC-MS enthalten) erhalten. Dieses Gemisch wurde direkt (ohne weitere Aufreinigung) in die betreffende Folgereaktion ein gesetzt (siehe Beispiel 203A).
LC-MS (Methode 2): Rt = 0.54 min; MS (ESIpos): m/z = 251 [M+H]+
Beispiel 110A
(+/-)-ie/f-Butyl-4-amino-3-(2-methoxyphenyl)-3-methylbutanoat (Racen
Eine Lösung aus (+/-)-fe/f-Butyl-3-cyan-3-(2-methoxyphenyl)butanoat (7.00 g, 25.4 mmol, Beispiel 40A) in ie f-Butanol (140 ml) wurde mit Raney-Nickel (1.49 g, 25.4 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert, die Mutterlauge eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 5.52 g (92% Reinheit, 72% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 0.63 min; MS (ESIpos): m/z = 280 [M+H]+
Beispiel 111 A
(+/-)-ie/f-Butyl-4-amino-3-(5-fluor-2-methoxyphenyl)butanoat-Hydrochlorid
Eine Lösung aus (+/-)-ie/f-Butyl-3-cyan-3-(5-fluor-2-methoxyphenyl)propanoat (150 mg, 537 μηηοΙ, Beispiel 41A) in Methanol (10 ml) unter Stickstoff wurde mit konzentrierter Salzsäure (150 μΙ, 12 M, 1.8 mmol), gefolgt von Platindioxid (29 mg, 83% Reinheit, 107 μηηοΙ) versetzt und anschließend 1 h bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert, die Mutterlauge eingeengt und der Rückstand im Vakuum getrocknet. In einem Wiederholexperiment wurde eine Lösung aus (+/-)-ierf-Butyl-3-cyan-3-(5-fluor-2-methoxyphenyl)propanoat (775 mg, 2.78 mmol, Beispiel 41A) in Methanol (50 ml) mit konzentrierter Salzsäure (0.55 ml, 12 M, 6.58 mmol), gefolgt von Platindioxid (106 mg, 83% Reinheit, 387 μηηοΙ) versetzt und 2 h bei Normal- druck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert, die Mutterlauge eingeengt und der Rückstand im Vakuum getrocknet. Die Rückstände aus beiden Experimenten wurden vereinigt und zwischen Dichlormethan und gesättigter Natriumhydrogencarbonat verteilt. Nach Phasentrennung wurde die wässrige Phase zweimal mit Dichlormethan extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rück- stand wurde mittels Flash-Säulenchromatographie (40 g Kieselgel Reveleris, Dichlormethan/(10% Methanol in Dichlormethan) 100:0— > 16:84) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und dreimal nacheinander mit ief-Butylmethylether versetzt und wieder eingeengt. Der Rückstand wurde in ierf-Butylmethylether gelöst und mit 1 M Salzsäure in Diethylether (1.94 ml, 1.94 mmol) versetzt und 30 min bei RT gerührt. Die Suspension wurde anschließend eingeengt, danach zweimal nacheinander mit ierf-Butylmethylether versetzt und wieder eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 541 mg (Reinheit 98%, 50% d. Th., bezogen auf die Summe an eingesetztem (+/-)-ierf-Butyl-3-cyan-3-(5-fluor-2-methoxyphenyl)propanoat) der Titelverbindung erhalten.
LC-MS (Methode 9): Rt = 2.35 min; MS (ESIpos): m/z = 284 [M-HCI+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.98 (br. s, 3H), 7.15-6.95 (m, 3H), 3.79 (s, 3H), 3.70- 3.60 (m, 1 H), 3.01 (br. s, 2H), 2.76 (dd, 1 H), 2.62 (dd, H), 1.22 (s, 9H).
Beispiel 112A
(+/-)-ie/f-Butyl-4-amino-3-(2-fluor-6-methoxyphenyl)butanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-3-cyan-3-(2-fluor-6-methoxyphenyl)propanoat (1.00 g, 93% Reinheit, 3.34 mmol, Beispiel 42A) in Methanol (30 ml) wurde mit Platindioxid (152 mg, 667 μηηοΙ) versetzt und 3 h bei Normaldruck hydriert. Anschließend wurde das Gemisch mit 1 M Salzsäure (3.3 ml, 3.3 mmol) versetzt und weitere 17 h bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert, und das Filtrat wurde mit gesättigter Natriumhyd- rogencarbonat-Lösung und Wasser (jeweils 30 ml) versetzt und zweimal mit Dichlormethan (jeweils 50 ml) extrahiert. Die vereinten organischen Phasen wurden über Magnesiumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 799 mg (82% Reinheit, 69% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.02 min; MS (ESIpos): m/z = 284 [M+H]+ Beispiel 113A
(+/-)-ie/f-Butyl-4-amino-3-(2-ethoxyphenyl)butanoat-Hydrochlorid (Racemat)

Eine Lösung aus (+/-)-ie/f-Butyl-3-cyan-3-(2-ethoxyphenyl)propanoat (150 mg, 537 μηηοΙ, Beispiel 43A) in Methanol (140 ml) unter Stickstoff wurde mit Platindioxid (384 mg, 83% Reinheit, 1.40 mmol) und konzentrierter Salzsäure (580 μΙ, 12 M, 7.0 mmol) versetzt und anschließend 4 h bei Normaldruck hydriert. Danach wurde der Katalysator abfiltriert, und das Filtrat wurde zwischen Dichlormethan und gesättigter Natriumhydrogencarbonat-Lösung verteilt. Nach Phasen- trennung wurde die wässrige Phase zweimal mit Dichlormethan extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet und filtriert. Das Filtrat wurde mit 2 M Hydrogenchlorid-Lösung in Diethylether (3.5 ml, 7.0 mmol) versetzt, eingeengt und anschließend mit Dichlormetan versetzt und wieder eingeengt. Der Rückstand wurde in Acetonitril gelöst, die Lösung wurde erneut eingeengt, und der Rückstand wurde im Vakuum getrocknet. Nach Ent- nähme von 200 mg des Rückstandes für Reinigungsversuche wurden anschließend 1.80 g (90% Reinheit, 73% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 10): Rt = 2.08 min; MS (ESpos): m/z = 280 [M-HCI+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.89 (br. s, 3H), 7.24 (dd, 1 H), 7.18 (d, 1 H), 6.95 (d, 1 H), 6.91 (dd, 1 H), 4.05 (q, 2H), 3.62-3.53 (m, 1 H), 3.04 (d, 2H), 2.75 (dd, 1 H), 2.66 (dd, 1 H), 1.38 (t, 3H, teilweise verdeckt), 1.20 (s, 9H).
Beispiel 114A
(+/-)-ie f-Butyl-4-amino-3-(5-fluor-2-methoxyphenyl)-3-methylbutanoat-Hydrochlorid (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-3-cyan-3-(5-fluor-2-methoxyphenyl)butanoat (1.38 g, 4.69 mmol, Beispiel 44A) in Methanol (95 ml) unter Stickstoff wurde mit konzentrierter Salzsäure (390 μΙ, 12 M, 4.7 mmol), gefolgt von Platindioxid (257 mg, 83% Reinheit, 938 μηηοΙ) versetzt und anschlie- ßend 2.5 h bei Normaldruck hydriert. Anschließend wurde der Katalysator abfiltriert, und das Filtrat wurde zwischen Dichlormethan und gesättigter Natriumhydrogencarbonat-Lösung verteilt. Nach Phasentrennung wurde die wässrige Phase zweimal mit Dichlormethan extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet und filtriert. Das Filtrat wurde anschließend mit 2 M Hydrogenchlorid-Lösung in Diethylether (2.3 ml, 4.7 mmol) versetzt, eingeengt und zweimal nacheinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde im Vakuum getrocknet. Es wurden 1.33 g (95% Reinheit, 81 % d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 1 ): Rt = 2.39 min; MS (ESpos): m/z = 298 [M-HCI+H]+
1H-NMR (400 MHz, CDCI3): δ [ppm] = 7.96 (br. s, 3H), 7.10-6.90 (m, 2H), 6.88-6.80 (m, 1 H), 3.86 (s, 3H), 3.48 (d, 1 H), 3.14 (d, 1 H), 3.03 (d, 1 H), 2.58 (d, 1 H), 1.62 (s, 3H), 1.22 (s, 9H).
Beispiel 115A
(+/-)-ierf-Butyl-4-amino-3-(3,5-difluor-2-methoxyphenyl)-3-methylbutanoat-Hydrochlorid (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-3-cyan-3-(3,5-difluor-2-methoxyphenyl)butanoat (1.54 g, 4.95 mmol, Beispiel 45A) in Methanol (100 ml) unter Stickstoff wurde mit Platindioxid-Hydrat (242 mg,
989 μηηοΙ), gefolgt von einer Lösung aus konzentrierter Salzsäure (410 μΙ, 12 M, 4.9 mmol) in Wasser (4.5 ml) versetzt und anschließend 4 h bei Normaldruck hydriert. Anschließend wurde der Katalysator abfiltriert, und das Filtrat wurde in zwei Portionen aufgeteilt und nacheinander jeweils mit gesättigter Natriumhydrogencarbonat-Lösung gewaschen und zweimal mit Dichlor- methan extrahiert. Die vereinigten organischen Phasen wurden jeweils über Natriumsulfat getrocknet und filtriert. Nach Zugabe von 1 M Hydrogenchlorid-Lösung in Diethylether (insgesamt 5 ml, anteilig aufgeteilt in zwei Portionen) wurde das erhaltene Gemisch jeweils eingeengt und der Rückstand jeweils im Vakuum getrocknet. Die beiden erhaltenen Rückstände wurden vereinigt. Es wurden 1.50 g (96% Reinheit, 83% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 1 ): Rt = 2.43 min; MS (ESpos): m/z = 316 [M-HCI+H]+
1H-NMR (400 MHz, CDCI3): δ [ppm] = 8.1 1 (br. s, 3H), 6.90-6.70 (m, 2H), 4.00 (s, 3H), 3.42 (dd, 1 H), 3.17 (dd, 1 H), 3.00 (d, 1 H), 2.60 (d, 1 H), 1.62 (s, 3H), 1.27 (s, 9H).
Beispiel 116A
(+/-)-ie/f-Butyl-4-amino-3-(4-fluor-2-methoxyphenyl)-3-methylbutanoat-Hydrochlorid (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-3-cyan-3-(4-fluor-2-methoxyphenyl)butanoat (3.02 g, 10.3 mmol, Beispiel 46A) in Methanol (200 ml) unter Stickstoff wurde mit Platindioxid-Hydrat (563 mg, 83% Reinheit, 2.06 mmol), gefolgt von konzentrierter Salzsäure (860 μΙ, 12 M, 10 mmol) versetzt und anschließend 4 h bei Normaldruck hydriert. Anschließend wurde der Katalysator abfiltriert, und das Filtrat wurde mit gesättigter Natriumhydrogencarbonat-Lösung gewaschen und zweimal mit Dichlormethan extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet und filtriert. Nach Zugabe von 2 M Hydrogenchlorid-Lösung in Diethylether (5.2 ml) wurde das erhaltene Gemisch eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 3.02 g (99% Reinheit, 87% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 1 ): Rt = 3.01 min; MS (ESpos): m/z = 298 [M-HCI+H]+
1H-NMR (400 MHz, CDC ): δ [ppm] = 7.90 (br. s, 3H), 7.20-7.15 (m, 1 H), 6.65-6.55 (m, 2H), 3.87 (s, 3H), 3.40 (m, 1 H), 3.13 (m, 1 H), 2.96 (d, 1 H), 2.58 (d, 1 H), 1.62 (s, 3H), 1.22 (s, 9H).
Beispiel 117A
(+/-)-Ethyl-2,2-difluor-3-(2-methoxyphenyl)-4-nitrobutanoat (Racemat)
In einem ausgeheizten Kolben unter Argon wurde eine aufgeschlämmten Rieke-Zink Suspension (ca. 5 ml, ca. 500 mg, ca. 7.7 mmol) auf 80 °C erhitzt und tropfenweise mit Ethyl-brom(difluor)- acetat (740 μΙ, 5.6 mmol) versetzt. Nach weiteren 5 min bei 80 °C wurde 1-Methoxy-2-[(£)-2- nitrovinyl]benzol (500 mg, 2.79 mmol) hinzugegeben, und es wurde weitere 15 min bei 80 °C gerührt. Anschließend wurde das Gemisch auf RT kommen gelassen und mit 1 M Salzsäure (ca. 20 ml) versetzt. Nachdem sich Reste von Zink aufgelöst hatten, wurde das Gemisch mit Ethylacetat extrahiert. Die organische Phase wurde mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels Flash- Säulenchromatographie (50 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat Gradient 1 :0— > 10:1 ) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert. Es wurden 105 mg (47% Reinheit, 3% d. Th.) der Titelverbindung erhalten. LC-MS (Methode 7): Rt = 1.31 min; MS (ESIneg): m/z = 302 [M-H]"
Beispiel 118A
(+/-)-Ethyl-4-amino-2,2-difluor-3-(2-methoxyphenyl)butanoat-Hydrochlorid (Racemat)
Ein Gemisch aus (+/-)-Ethyl-2,2-difluor-3-(2-methoxyphenyl)-4-nitrobutanoat (95 mg, 313 μηηοΙ, nicht reinheitskorrigiert, Beispiel 1 17A) in einem Gemisch aus Methanol (3 ml) und 1 M Salzsäure (630 μΙ) wurde mit Platindioxid (30 mg, 132 μηηοΙ) versetzt und über Nacht hydriert. Anschließend wurden erneut Platindioxid (30 mg, 132 μηηοΙ) und 1 M Salzsäure (1 ml) hinzugegeben, und das Gemisch wurde weitere 3 Tage bei RT hydriert. Anschließend wurde das Gemisch über Kieselgur filtriert und eingeengt. Der Rückstand wurde mehrere Male hintereinander mit Dich- lormethan versetzt und wieder eingeengt. Nach Trocknen im Vakuum wurden 80 mg (35% Reinheit, 33% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 7): Rt = 0.72 min; MS (ESIpos): m/z = 274 [M+H]+ Beispiel 119A
(+/-)-ie/f-Butyl-4-amino-3-(2-fluor-6-methoxyphenyl)-3-methylbutanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-3-cyan-3-(2-fluor-6-methoxyphenyl)butanoat (798 mg, 79% Reinheit, 2.14 mmol, Beispiel 47A) in Methanol (19 ml) wurde mit Platindioxid (97.0 mg, 427 μηηοΙ), gefolgt von 1 M Salzsäure (2.1 ml, 2.1 mmol) versetzt und anschließend 17 h bei Normaldruck hydriert. Anschließend wurde der Katalysator abfiltriert, und das Filtrat wurde mit gesättigter Natriumhydrogencarbonat-Lösung und Wasser (jeweils 30 ml) versetzt und zweimal mit Dichlormethan (jeweils 50 ml) extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 714 mg (82% Reinheit, 92% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 0.66 min; MS (ESIpos): m/z = 298 (M+H)+
Beispiel 120A
ie/f-Butyl-4-amino-3-(2-chlorphenyl)pentanoat (Diastereomerengemisch)
Eine Lösung aus ie/f-Butyl-3-(2-chlorphenyl)-4-nitropentanoat (602 mg, 1.92 mmol, Beispiel 48A) in Ethanol (30 ml) wurde bei RT mit 1 M Salzsäure (26.1 ml, 26.1 mmol) und Zinkstaub (5.01 g, 76.74 mmol) versetzt und einen Tag bei RT gerührt. Anschließend wurde das Gemisch mit ge- sättigter Natriumhydrogencarbonat-Lösung (ca. 50 ml) auf pH 9 eingestellt, mit Wasser (50 ml) und Ethylacetat (100 ml) versetzt und geschüttelt. Der vorhandene Feststoff wurde über Kieselgur abfiltriert. Das Filtrat wurde in organische und wässrige Phase getrennt, und die wässrige Phase wurde einmal mit Ethylacetat (50 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung (100 ml) gewaschen und über Natri-
umsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde kurz im Vakuum getrocknet. Es wurden 493 mg (74% Reinheit, 67% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 0.61 & 0.63 min; MS (ESIpos): jeweils m/z = 284 (M+H)+ Beispiel 121A
(+/-)-fe/f-Butyl-5-amino-4-(2-chlorphenyl)pentanoat (Racemat)
Eine Lösung aus (+/-)-fe/f-Butyl-4-(2-chlorphenyl)-4-cyanbutanoat (4.50 g, 16.1 mmol, Beispiel 49A) in ie f-Butanol (90 ml) wurde mit Raney-Nickel (944 mg, 16.1 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde das Gemisch erneut mit Raney-Nickel (944 mg, 16.1 mmol) versetzt und weitere 24 h bei Normaldruck hydriert. Danach wurde der Katalysator über Kieselgur abfiltriert und die Mutterlauge eingeengt. Der Rückstand wurde in Dichlorme- than aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap- Cartridge KP-Sil, Cyclohexan/Ethylacetat-Gradient 93:7— > 7:3, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 1.62 g (91% Reinheit, 32% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 4): Rt = 1.70 min; MS (ESIpos): m/z = 284 [M+H]+ Beispiel 122A
(+/-)-ie/f-Butyl-5-amino-4-(2-chlorphenyl)-4-methylpentanoat (Racemat)
Eine Lösung aus (+/-)-fe/f-Butyl-4-(2-chlorphenyl)-4-cyanpentanoat (1.50 g, 5.1 1 mmol, Beispiel 50A) in ie f-Butanol (30 ml) wurde mit Raney-Nickel (300 mg, 5.1 1 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde das Gemisch erneut mit Raney-Nickel (944 mg, 16.1 mmol) versetzt und weitere 24 h bei Normaldruck hydriert. Anschließend wurde das Gemisch ein weiteres mal mit Raney-Nickel (944 mg, 16.1 mmol) versetzt und weitere 24 h bei Normaldruck hydriert. Danach wurde der Katalysator über Kieselgur abfiltriert und die Mutterlau-
ge eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 840 mg (73% Reinheit, 40% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.22 min; MS (ESIpos): m/z = 298 [M+H]+ Beispiel 123A
(+/-)-ie/f-Butyl-5-amino-4-(2-chlor-6-fluorphenyl)pentanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-4-(2-chlor-6-fluorphenyl)-4-cyanbutanoat (1.45 g, 4.87 mmol, Beispiel 51A) in ie f-Butanol (30 ml) wurde mit Raney-Nickel (286 mg, 4.87 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde das Gemisch erneut mit Raney-Nickel (286 mg, 4.87 mmol) versetzt und weitere 24 h bei Normaldruck hydriert. Danach wurde der Katalysator über Kieselgur abfiltriert und die Mutterlauge eingeengt. Es wurden 1.28 g (67% Reinheit, 58% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.19 min; MS (ESIpos): m/z = 302 [M+H]+ Beispiel 124A
(+/-)-ie/f-Butyl-5-amino-4-(2-methylphenyl)pentanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-4-cyan-4-(2-methylphenyl)butanoat (534 mg, 2.06 mmol, Beispiel 52A) in ie f-Butanol (13 ml) wurde mit Raney-Nickel (121 mg, 2.06 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert und die Mutterlauge eingeengt. Es wurden 505 mg (95% Reinheit, 89% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.18 min; MS (ESIpos): m/z = 264 [M+H]+
Beispiel 125A
(+/-)-ie/f-Butyl-5-amino-4-(2-chlor-5-fluorphenyl)pentanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-4-(2-chlor-5-fluorphenyl)-4-cyanbutanoat (14.0 g, 93% Reinheit, 43.7 mmol, Beispiel 53A) in ief-Butanol (260 ml) wurde mit Raney-Nickel (2.57 g, 43.7 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde das Gemisch erneut mit Raney-Nickel (2.57 g, 43.7 mmol) versetzt und weitere 24 h bei Normaldruck hydriert. Anschließend wurde das Gemisch ein weiteres mal mit Raney-Nickel (2.57 g, 43.7 mmol) versetzt und weitere 24 h bei Normaldruck hydriert. Danach wurde der Katalysator über Kieselgur abfiltriert und die Mutterlauge eingeengt. Es wurden 14.4 g (60% Reinheit, 65% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.14 min; MS (ESIpos): m/z = 302 [M+H]+ Beispiel 126A
(+/-)-ie/f-Butyl-5-amino-4-(2-methoxyphenyl)pentanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-4-cyan-4-(2-methoxyphenyl)butanoat (495 mg, 1.80 mmol, Beispiel 54A) in ie f-Butanol (1 1 ml) wurde mit Raney-Nickel (106 mg, 1.80 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert und die Mutterlauge eingeengt. Es wurden 269 mg (85% Reinheit, 46% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 0.68 min; MS (ESIpos): m/z = 280 [M+H] Beispiel 127A
(+/-)-ie/f-Butyl-5-amino-4-(2-chlor-5-fluorphenyl)-4-methylpentanoat (Racemat)
Eine Lösung aus (+/-)-fe/f-Butyl-4-(2-chlor-5-fluorphenyl)-4-cyanpentanoat (1.20 g, 3.85 mmol, Beispiel 55A) in fe/f-Butanol (25 ml) wurde mit Raney-Nickel (226 mg, 3.85 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfil- triert und die Mutterlauge eingeengt. Es wurden 1.54 g (66% Reinheit, 84% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.24 min; MS (ESIpos): m/z = 316 [M+H]+ Beispiel 128A
(+/-)-ie/f-Butyl-5-amino-4-(2-chlor-3,6-difluorphenyl)pentanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-4-(2-chlor-3,6-difluorphenyl)-4-cyanbutanoat (4.10 g, 13.0 mmol, Beispiel 56A) in ie f-Butanol (75 ml) wurde mit Raney-Nickel (762 mg, 13.0 mmol) versetzt und 24 h bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert, zweimal mit ie f-Butanol (15 ml) nachgewaschen und die Mutterlauge eingeengt. Der Rückstand wurde in Ethylacetat (80 ml) aufgenommen und die Lösung nacheinander mit 1 M Salzsäure und Wasser (jeweils 80 ml) extrahiert. Die vereinigten wässrigen Phasen wurden mit gesättigter Nat- riumhydrogencarbonat-Lösung auf pH 8-9 eingestellt und zweimal mit Ethylacetat (jeweils 100 ml) extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 1.76 g (100% Reinheit, 42% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.21 min; MS (ESIpos): m/z = 320 [M+H]+
Beispiel 129A
(+/-)-ie/f-Butyl-5-amino-4-(2-fluorphenyl)pentanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-4-cyan-4-(2-fluorphenyl)butanoat (4.22 g, 16.0 mmol, nicht rein- heitskorrigiert, Beispiel 57A) in ie f-Butanol (100 ml) wurde mit Raney-Nickel (941 mg, 16.0 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde erneut mit Raney- Nickel (941 mg, 16.0 mmol) versetzt und eine weitere Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert und die Mutterlauge eingeengt. Es wurden 3.02 g (60% Reinheit, 42% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 0.61 min; MS (ESIpos): m/z = 268 [M+H]+
Beispiel 130A
(+/-)-ie/f-Butyl-5-amino-4-[2-(difluormethoxy)phenyl]pentanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-4-cyan-4-[2-(difluormethoxy)phenyl]butanoat (3.85 g, 12.4 mmol, Beispiel 58A) in ie f-Butanol (75 ml) wurde mit Raney-Nickel (726 mg, 12.4 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfil- triert und die Mutterlauge eingeengt. Es wurden 2.92 g (80% Reinheit, 60% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 0.68 min; MS (ESIpos): m/z = 316 [M+H]+ Beispiel 131A
(+/-)-ie/f-Butyl-5-amino-4-(2,6-difluorphenyl)pentanoat (Racemat)
Eine Lösung aus (+/-)-fe/f-Butyl-4-cyan-4-(2,6-difluorphenyl)butanoat (4.80 g, 17.1 mmol, Beispiel 59A) in ie f-Butanol (100 ml) wurde mit Raney-Nickel (1.00 g, 17.1 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert und die Mutterlauge eingeengt. Es wurden 4.06 g (81 % Reinheit, 67% d. Th.) der Titelverbin- dung erhalten.
LC-MS (Methode 2): Rt = 0.64 min; MS (ESIpos): m/z = 286 [M+H]+ Beispiel 132A
(+/-)-ie/f-Butyl-5-amino-4-(2-chlor-3-fluorphenyl)pentanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-4-(2-chlor-3-fluorphenyl)-4-cyanbutanoat (4.32 g, 14.5 mmol, Beispiel 60A) in ie/f-Butanol (85 ml) wurde mit Raney-Nickel (852 mg, 14.5 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert, zweimal mit fe/f-Butanol (15 ml) nachgewaschen und die Mutterlauge eingeengt. Der Rückstand wurde in Ethylacetat (80 ml) aufgenommen und nacheinander mit 1 M Salzsäure und Wasser (jeweils 80 ml) extrahiert. Die vereinigten wässrigen Phasen wurden mit gesättigter Nat- riumhydrogencarbonat-Lösung auf pH 8-9 eingestellt und zweimal mit Ethylacetat (80 ml) extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 2.43 g (85% Reinheit, 47% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.28 min; MS (ESIpos): m/z = 302 [M+H]+ Beispiel 133A
(+/-)-ie/f-Butyl-/V-[2-amino-1-(2-chlorphenyl)ethyl]-/V-methylglycinat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-/V-[(2-chlorphenyl)(cyan)methyl]-/V-methylglycinat (1.40 g, 4.75 mmol, Beispiel 61A) in ie/f-Butanol (28 ml) wurde mit Raney-Nickel (279 mg, 4.75 mmol)
versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde erneut Raney-Nickel (279 mg, 4.75 mmol) hinzugegeben, und das Gemisch wurde weitere 24 h bei Normalduck hydriert. Danach wurde der Katalysator über Kieselgur abfiltriert und die Mutterlauge eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 800 mg (41 % Reinheit, 23% d. Th.) der Titel- Verbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.21 min; MS (ESIpos): m/z = 299 [M+H]+ Beispiel 134A
(+/-)-ie/f-Butyl-/V-[2-amino-1-(2-chlorphenyl)ethyl]glycinat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-/V-[(2-chlorphenyl)(cyan)methyl]glycinat (1.40 g, „4.99 mmol", nicht reinheitskorrigiert, Beispiel 62A) in ie/f-Butanol (29 ml) wurde mit Raney-Nickel (293 mg, 4.99 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert und die Mutterlauge eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 1.25 g (ca. 50% Reinheit, 44% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 0.66 min; MS (ESIpos): m/z = 285 [M+H]+ Beispiel 135A
(+/-)-ie/f-Butyl-[2-(2-chlorphenyl)-2-hydroxyethyl]carbamat (Racemat)
Zu einer Lösung aus (+/-)-2-Amino-1-(2-chlorphenyl)ethanol (3.98 g, 23.2 mmol) in Dichlorme- than (50 ml) wurden bei RT Triethylamin (3.6 ml, 26 mmol) und Di-ie/f-butyldicarbonat (5.67 g, 26.0 mmol) gegeben (Gasentwicklung), und das Gemisch wurde 18 h bei RT gerührt. Anschließend wurde das Gemisch mit Dichlormethan (100 ml) verdünnt, mit verdünnter Natriumhydro- gencarbonat-Lösung und gesättigter Natriumchlorid-Lösung (jeweils 100 ml) gewaschen, über
Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 7.50 g (53% Reinheit laut GC-MS, 63% d. Th.) der Titelverbindung erhalten.
GC-MS (Methode 12): Rt = 6.53 min, MS (Elpos): m/z = 215 [M-C4H8]+ Beispiel 136A
(+/-)-Ethyl-[2-[(ie/f-butoxycarbonyl)amino]-1 -(2-chlorphenyl)ethoxy]acetat (Racemat)
Zu einer Lösung aus (+/-)-ie/f-Butyl-[2-(2-chlorphenyl)-2-hydroxyethyl]carbamat (7.41 g, 85% Reinheit, 23.2 mmol, Beispiel 135A), in Dichlorethan (50 ml) unter Argon wurden Ethyl-diazoacetat (4.6 ml, 44 mmol) und Rhodium(ll)acetat-Dimer bei RT gegeben (Gasentwicklung), und das Ge- misch wurde 17 h bei RT gerührt. Anschließend wurde erneut Ethyl-diazoacetat (4.6 ml, 44 mmol) hinzugegeben, und das Gemisch wurde weitere vier Tage bei RT gerührt und anschließend unter Bildung eines Rückstandes („Rückstand 1") eingeengt. Ein Teil dieses„Rückstandes 1" (ca. 1.4 g) wurde in Dichlormethan (50 ml) gelöst, und die Lösung wurde mit Wasser (50 ml) und verdünnter Natriumhydrogencarbonat-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und einge- engt. Der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (50 g Kieselgel Biotage Snap-Cartridge Ultra, Cyclohexan/Ethylacetat 97:3— > 7:3, Isolera One) gereinigt. Nach Einengen und Trocknen der vereinigten Zielfraktionen wurden 300 mg (75% Reinheit, 3% d. Th.) einer ersten Charge der Titelverbindung erhalten. Die restliche Menge des „Rückstandes 1 " (ca. 12 g) wurde in Dichlormethan aufgenommen und in zwei Portionen mittels Flash-Säulenchromatographie (jeweils 100 g Kieselgel Biotage Snap-Cartridge Ultra, Cyclohexan/Ethylacetat 97:3— > 7:3, Isolera One) gereinigt. Nach Einengen und Trocknen der vereinigten Zielfraktionen wurden 2.89 g (86% Reinheit, 31% d. Th.) einer zweiten Charge der Titelverbindung erhalten. Von dieser zweiten Charge wurden 500 mg entnommen, in Acetonitril gelöst und mittels präparativer HPLC (Methode 16) nachgereinigt. Die Zielfraktionen wurden vereinigt und mit gesät- tigter Natriumhydrogencarbonat-Lösung auf pH 7-8 eingestellt. Die Acetonitril-Phase wurde am Rotationsverdampfer entfernt, und die wässrige Phase wurde zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 234 mg (100% Reinheit) der nachgereinigten Titelverbindung (siehe Analytik) erhalten.
LC-MS (Methode 1 ): Rt = 2.13 min; MS (ESIpos): m/z = 358 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.150 (0.1 1 ), 0.007 (0.79), 1.150 (3.98), 1.168 (8.32), 1.186 (4.12), 1.254 (1.35), 1.332 (16.00), 1.987 (1.08), 2.327 (0.14), 2.365 (0.15), 2.669 (0.14), 2.709 (0.15), 3.213 (1.08), 3.223 (1.17), 3.238 (0.75), 3.91 1 (1.58), 3.952 (2.73), 4.020 (0.28), 4.043 (1.39), 4.067 (1.23), 4.085 (3.99), 4.102 (3.21 ), 4.120 (1.04), 4.905 (0.64), 4.920 (0.93), 4.934 (0.49), 6.774 (0.68), 7.310 (0.28), 7.329 (0.82), 7.348 (0.84), 7.361 (0.83), 7.379 (1.24), 7.395 (0.57), 7.418 (1.21 ), 7.444 (1.55), 7.463 (0.85).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 7.51-7.24 (m, 4H), 6.78 (t, 1 H), 4.92 (t, 1 H), 4.09 (q, 2H), 4.06-3.86 (m, 2H), 3.28-3.16 (m, 2H), 1.33 (s, 7H), 1.26 (br. s, 2H), 1.17 (t, 3H).
Beispiel 137A
(+/-)-Ethyl-[2-amino-1-(2-chlorphenyl)ethoxy]acetat-Hydrochlorid (Racen
(+/-)-Ethyl-[2-[(ie/f-butoxycarbonyl)amino]-1-(2-chlorphenyl)ethoxy]acetat (1.91 g, 5.34 mmol, Beispiel 136A) wurde in einer 4 M Lösung aus Hydrogenchlorid in Dioxan gelöst und 2.5 h bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlorme- than versetzt und wieder eingeengt. Der Rückstand wurde im Vakuum getrocknet. Es wurden 1.90 g (72% Reinheit, 87% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 0.52 min; MS (ESIpos): m/z = 258 [M+H]+ Beispiel 138A
(+/-)-Methyl-{[2-amino-1 -(2-chlorphenyl)ethyl]sulfanyl}acetat (Racemat)
Eine Lösung aus (+/-)-Methyl-{[1-(2-chlorphenyl)-2-nitroethyl]sulfanyl}acetat (1.40 g, 4.83 mmol, Beispiel 63A) in Methanol (28 ml) und Essigsäure (14 ml) wurde bei RT mit Zinn(ll)chlorid versetzt, und das Gemisch wurde 3 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das Gemisch eingeengt, und der Rückstand wurde in Acetonitril gelöst und mittels praparativer
HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 3.80 g (50% Reinheit laut HPLC-MS, „>100% d. Th.", lösungsmittelhaltig) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 0.79 min; MS (ESIpos): m/z = 260 [M+H]+ Beispiel 139A
(+/-)-Methyl-({2-[(ie/f-butoxycarbonyl)amino]-1-(2-chlorphenyl)ethyl}sulfanyl)acetat (Racemat)
Eine Lösung aus (+/-)-Methyl-{[2-amino-1-(2-chlorphenyl)ethyl]sulfanyl}acetat (1.56 g, 45% Reinheit, 2.70 mmol, Beispiel 138A) in Dichlormethan (5.8 ml) wurde bei RT nacheinander mit Triethylamin (420 μΙ, 3.0 mmol) und Di-ie/f-butyldicarbonat (661 mg, 3.03 mmol) versetzt (Gasentwicklung), und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch mit Dichlormethan auf ein Volumen von 100 ml verdünnt und einmal mit halbkonzentrierter Natriumhydrogencarbonat-Lösung gewaschen. Der dabei entstandene Niederschlag wurde abfiltriert und verworfen. Danach wurde das Gemisch mit gesättigter wässriger Natriumchlorid- Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde mittels praparativer HPLC (Methode 16) zweimal hintereinander gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 100 mg (98% Reinheit, 10% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.00 min; MS (ESIpos): m/z = 382 [M+Na]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 7.50-7.41 (m, 2H), 7.40-7.23 (m, 2H), 6.99 (t, 1 H), 4.60 (t, 1 H), 3.55 (s, 3H), 3.50-3.28 (m, 3H), 3.23-3.13 (m, 1 H), 1.33 (s, 9H).
Beispiel 140A
(+/-)-Methyl-{[2-[(ie/f-butoxycarbonyl)amino]-1-(2-chlorphenyl)ethyl]sulfonyl}acetat (Racemat)
Eine Lösung aus (+/-)-Methyl-({2-[(ierf-butoxycarbonyl)amino]-1-(2-chlorphenyl)ethyl}sulfanyl)- acetat (100 mg, 278 μηηοΙ, Beispiel 139A) in Dichlormethan (5 ml) wurde bei RT mit meta- Chlorperbenzoesaeure (103 mg, 70% Reinheit, 417 μηηοΙ) versetzt, und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch mit gesättigter Natriumhydrogencarbo- nat-Lösung (5 ml) versetzt, kräftig verrührt und danach mit Dichlormethan und Wasser versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Dichlormethan extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Acetonitril gelöst und mittels präparativer HPLC (Methode 17) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 75 mg (100% Reinheit, 69% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.77 min; MS (ESIneg): m/z = 390 [M-H]" Beispiel 141A
(+/-)-Methyl-{[2-amino-1-(2-chlorphenyl)ethyl]sulfonyl}acetat-Hydrochlorid
(+/-)-Methyl-{[2-[(ie/f-butoxycarbonyl)amino]-1-(2-chlorphenyl)ethyl]sulfonyl}acetat (75 mg, 191 μηηοΙ, Beispiel 140A) wurde in einer 4 M Lösung aus Hydrogenchlorid in Dioxan (720 μΙ, 2.9 mmol) gelöst und 3 h bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Es wurden 60 mg (97% Reinheit, 93% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 4): Rt = 1.03 min; MS (ESIpos): m/z = 292 [M-HCI+H]+
Beispiel 142A
(+/-)-ie/f-Butyl-5-amino-4-(6-chlor-2,3-difluorphenyl)pentanoat (Racemat)
N H2 O CH3
Eine Lösung aus (+/-)-ie/f-Butyl-4-(6-chlor-2,3-difluorphenyl)-4-cyanbutanoat (2.26 g, 83% Reinheit, 5.95 mmol, Beispiel 64A) in ie f-Butanol (35 ml) wurde mit Raney-Nickel (349 mg, 5.95 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert, zweimal mit ie f-Butanol (10 ml) nachgewaschen und das Filtrat eingeengt. Der Rückstand wurde in Ethylacetat (50 ml) aufgenommen und nacheinander mit 1 M Salzsäure und Wasser (jeweils 50 ml) extrahiert. Die vereinigten wässrigen Phasen wurden mit gesättigter Natriumhydrogencarbonat-Lösung auf pH 8-9 eingestellt und zweimal mit Ethylacetat (50 ml) extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 1.05 g (97% Reinheit, 53% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 4): Rt = 1.82 min; MS (ESIpos): m/z = 320 [M+H]+ Beispiel 143A
(+/-)-Methyl-5-amino-4-[2-(trifluormethyl)phenyl]pentanoat (Racemat)
Eine Lösung aus (+/-)-Methyl-4-cyan-4-[2-(trifluormethyl)phenyl]butanoat (4.40 g, 16.2 mmol, Beispiel 65A) in Methanol (150 ml) wurde mit Platindioxid (737 mg, 3.24 mmol) und konzentrierter Salzsäure (1.4 ml, 12 M, 16 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert, mit Methanol nachgewaschen, und das Filt- rat wurde eingeengt. Der Rückstand wurde mit dem aus einem Vorversuch analog erhaltenen Rückstand (eingesetzte Menge an (+/-)-Methyl-4-cyan-4-[2-(trifluormethyl)phenyl]butanoat: 200 mg, 737 μηηοΙ) vereinigt. Es wurden 5.50 g (77% Reinheit, 91 % d. Th., bezogen auf insgesamt 16.94 mmol eingesetztem (+/-)-Methyl-4-cyan-4-[2-(trifluormethyl)phenyl]butanoat) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 0.52 min; MS (ESIpos): m/z = 276 [M+H]+
Beispiel 144A
(+/-)-ie/f-Butyl-5-amino-4-(5-fluor-2-methylphenyl)pentanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-4-cyan-4-(5-fluor-2-methylphenyl)butanoat (4.92 g, 17.7 mmol, Beispiel 66A) in ierf-Butanol (100 ml) wurde mit Raney-Nickel (1.04 g, 17.7 mmol) versetzt und 24 h bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert, zweimal mit ierf-Butanol (15 ml) nachgewaschen und das Filtrat eingeengt. Der Rückstand wurde in Ethyl- acetat (80 ml) aufgenommen und nacheinander mit 1 M Salzsäure und Wasser (jeweils 80 ml) extrahiert. Die vereinigten wässrigen Phasen wurden mit gesättigter Natriumhydrogencarbonat-Lö- sung auf pH 8-9 eingestellt und zweimal mit Ethylacetat (80 ml) extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 2.04 g (92% Reinheit, 38% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 4): Rt = 1.94 min; MS (ESIpos): m/z = 282 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.109 (0.34), 1.197 (0.07), 1.293 (0.03), 1.357 (16.00), 1.394 (0.15), 1.513 (0.24), 1.609 (0.1 1 ), 1.621 (0.1 1 ), 1.638 (0.25), 1.648 (0.17), 1.663 (0.22), 1.681 (0.14), 1.709 (0.06), 1.927 (0.05), 1.943 (0.10), 1.950 (0.13), 1.964 (0.28), 1.986 (1.50), 1.995 (1.06), 2.01 1 (0.26), 2.018 (0.20), 2.072 (0.03), 2.232 (3.57), 2.303 (0.20), 2.366 (0.03), 2.641 (0.07), 2.659 (0.1 1 ), 2.672 (0.57), 2.681 (0.59), 2.690 (0.75), 2.696 (0.71 ), 2.71 1 (0.09), 2.728 (0.08), 2.851 (0.21 ), 2.863 (0.20), 3.172 (0.05), 3.312 (0.65), 6.866 (0.17), 6.873 (0.21 ), 6.887 (0.37), 6.894 (0.43), 6.909 (0.20), 6.916 (0.23), 6.968 (0.43), 6.974 (0.37), 6.994 (0.42), 7.001 (0.36), 7.105 (0.03), 7.150 (0.35), 7.166 (0.40), 7.187 (0.30).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 7.17 (dd, 1 H), 6.99 (dd, 1 H), 6.89 (td, 1 H), 2.93-2.80 (m, 1 H), 2.74-2.62 (m, 2H), 2.23 (s, 3H), 2.06-1.87 (m, 3H), 1.74-1.60 (m, 1 H), 1.50 (br. s, 2H), 1.36 (s, 9H).
Beispiel 145A
(+/-)-ie/f-Butyl-5-amino-4-[2-(trifluormethoxy)phenyl]pentanoat (Racemat)
N H2 O C H3
Eine Lösung aus (+/-)-ierf-Butyl-4-cyan-4-[2-(trifluormethoxy)phenyl]butanoat (3.13 g, 80% Reinheit, 7.59 mmol, Beispiel 67A) in ief-Butanol (45 ml) wurde mit Raney-Nickel (446 mg, 7.59 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde erneut Raney-Nickel (446 mg, 7.59 mmol) hinzugegeben, und das Gemisch wurde weitere 24 h bei Normaldruck hydriert. Danach wurde der Katalysator über Kieselgur abfiltriert und das Filtrat eingeengt und im Vakuum getrocknet. Es wurden 3.00 g (83% Reinheit, 98% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.29 min; MS (ESIpos): m/z = 334 [M+H]+ Beispiel 146A
(+/-)-ie/f-Butyl-5-amino-4-(pyridin-2-yl)pentanoat (Racemat)
(+/-)-ie/f-Butyl-4-cyan-4-(pyridin-2-yl)butanoat (799 mg, 3.24 mml, Beispiel 68A) wurde in ie/f- Butanol (10 ml) gelöst, mit Raney-Nickel (228 mg, 3.89 mmol) versetzt, und das Reaktionsgemisch wurde bei Normaldruck unter Wasserstoffatmosphäre über Nacht gerührt. Das Reaktions- gemisch wurde filtriert und eingeengt. Es wurden 678 mg der Titelverbindung als Rohprodukt erhalten. Das Rohprodukt wurde direkt (ohne weitere Reinigung) in die Folgereaktion eingesetzt.
LC-MS (Methode 6): Rt = 1.08 min; MS (ESIpos): m/z = 251 [M+H]+ Beispiel 147A
(+/-)-ie/f-Butyl-5-amino-4-[2-fluor-6-(trifluormethyl)phenyl]pentanoat (Racemat)

Eine Lösung von (+/-)-ie/f-Butyl-4-cyan-4-[2-fluor-6-(trifluormethyl)phenyl]butanoat (4.52 g, 13.6 mmol, Beispiel 69A) in ie/f-Butanol (100 ml) und Methanol (15 ml) wurde mit Raney-Nickel (801 mg, 13.6 mmol) versetzt und das Gemisch über Nacht unter Rühren bei Normaldruck hydriert. Das Raktionsgemisch wurde noch einmal mit Raney-Nickel (2 g, 34.0 mmol) versetzt und 40 h unter Normaldruck Wasserstoff stark gerührt. Anschließend wurde der Katalysator über Kieselgur abfiltriert, dreimal mit Methanol (je 30 ml) nachgewaschen und das Filtrat in Vakuum eingeengt. Der Rückstand wurde in 200 ml Ethylacetat aufgenommen. Diese organische Phase wurde zweimal mit 200 ml 1 M Salzsäure extrahiert. Die vereinigten wäßrigen Phasen wurden durch langsame Zugabe von Natriumhydrogencarbonat auf pH 8-9 gebracht, dann zweimal mit je 200 ml Ethylacetat extrahiert. Die vereinten organischen Phasen wurden über Natriumsulfat getrocknet und am Rotationsverdampfer eingeengt. Der Rückstand wurde im Vakuum getrocknet. Es wurden 3.32 g (92% Reinheit, 67% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.21 min; MS (ESIpos): m/z = 336 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.109 (0.25), 1.156 (0.06), 1.167 (0.07), 1.174 (0.12), 1.191 (0.06), 1.327 (16.00), 1.390 (0.40), 1.484 (0.07), 1.882 (0.09), 1.905 (0.14), 1.930 (0.25), 1.946 (0.23), 1.965 (0.29), 1.979 (0.15), 1.987 (0.45), 2.006 (0.19), 2.034 (0.10), 2.055 (0.30), 2.074 (0.35), 2.093 (0.31 ), 2.118 (0.16), 2.129 (0.1 1 ), 2.150 (0.04), 2.310 (0.07), 2.365 (0.02), 2.669 (0.02), 2.782 (0.10), 2.799 (0.17), 2.812 (0.24), 2.828 (0.25), 2.872 (0.18), 2.876 (0.18), 2.890 (0.29), 2.895 (0.30), 2.920 (0.37), 3.494 (0.02), 3.522 (0.03), 4.019 (0.05), 4.037 (0.05), 7.476 (0.66), 7.485 (0.49), 7.489 (0.49), 7.501 (0.31 ), 7.509 (0.87), 7.521 (0.09), 7.550 (0.51 ), 7.557 (0.32), 7.564 (0.37), 7.573 (0.26), 7.614 (0.04), 7.637 (0.04).
Beispiel 148A
(+/-)-ie/f-Butyl-5-amino-4-(2,3,5,6-tetrafluorphenyl)pentanoat (Racemat)
Eine Lösung von (+/-)-ie/f-Butyl-4-cyan-4-(2,3,5,6-tetrafluorphenyl)butanoat (850 mg, 2.68 mmol, Beispiel 70A) in ie f-Butanol (20 ml) und Methanol (2.1 ml) wurde mit Raney-Nickel (157 mg, 2.68 mmol) versetzt, und das Gemisch wurde über Nacht unter Rühren bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert, zweimal mit ierf-Butanol (10 ml) und Methanol (10 ml) nachgewaschen und das Filtrat im Vakuum eingeengt. Der Rückstand wurde in Ethylacetat (100 ml) aufgenommen. Diese organische Phase wurde zweimal mit 1 M Salzsäure (100 ml) extrahiert. Die vereinigten wässrigen Phasen wurden durch langsame Zuga-
be von Natriumhydrogencarbonat auf pH 8-9 gebracht und dann zweimal mit Ethylacetat (jeweils 100 ml) extrahiert. Die vereinten organischen Phasen wurden über Natriumsulfat getrocknet und am Rotationsverdampfer eingeengt. Es wurden 586 mg (95% Reinheit, 65% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.15 min; MS (ESIpos): m/z = 322 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.219 (0.06), 1.379 (16.00), 1.536 (0.07), 2.073 (0.16), 2.091 (0.22), 2.107 (0.28), 2.125 (0.26), 2.144 (0.10), 2.213 (0.07), 2.231 (0.19), 2.250 (0.22), 2.267 (0.18), 2.285 (0.13), 2.302 (0.08), 2.327 (0.07), 2.365 (0.07), 2.384 (0.36), 2.393 (0.38), 2.402 (0.59), 2.410 (0.56), 2.420 (0.29), 2.428 (0.27), 2.452 (0.06), 2.669 (0.06), 4.373 (0.20), 4.390 (0.39), 4.407 (0.19), 7.698 (0.31 ), 7.71 1 (1.11 ), 7.722 (0.54), 7.748 (0.35), 7.766 (0.1 1 ).
Beispiel 149A
(+/-)-ie/f-Butyl-5-amino-4-(2,3,5-trifluorphenyl)pentanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-4-cyan-4-(2,3,5-trifluorphenyl)butanoat (600 mg, 2.00 mmol, Beispiel 71 A) in ie/f-Butanol (15 ml) wurde mit Raney-Nickel (1 18 mg, 2.00 mmol) versetzt und über Nacht bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert, zweimal mit ie f-Butanol (je 5 ml) nachgewaschen und das Filtrat im Vakuum eingeengt. Der Rückstand wurde in Ethylacetat (50 ml) aufgenommen. Diese organische Phase wurde zweimal mit 1 M Salzsäure (jeweils 50 ml) extrahiert. Die vereinigten wässrigen Phasen wurden durch langsame Zugabe von Natriumhydrogencarbonat auf pH 8-9 gebracht und dann zweimal mit Ethylacetat (jeweils 50 ml) extrahiert. Die vereinten organischen Phasen wurden über Natriumsulfat getrocknet und am Rotationsverdampfer eingeengt. Es wurden 200 mg (96% Reinheit, 32% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 3): Rt = 1.81 min; MS (ESIpos): m/z = 304 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.1 10 (0.34), 1.193 (0.08), 1.234 (0.08), 1.353 (16.00), 1.509 (0.17), 1.639 (0.07), 1.673 (0.16), 1.689 (0.20), 1.698 (0.17), 1.714 (0.17), 1.729 (0.09), 1.865 (0.06), 1.934 (0.06), 1.955 (0.13), 1.968 (0.17), 1.984 (0.18), 2.003 (0.18), 2.025 (0.88), 2.041 (0.91 ), 2.059 (0.29), 2.328 (0.05), 2.367 (0.04), 2.691 (0.14), 2.710 (0.21 ), 2.723 (0.39), 2.742 (0.46), 2.755 (0.40), 2.771 (0.46), 2.787 (0.14), 2.802 (0.16), 2.944 (0.18), 7.039 (0.22),
7.051 (0.22), 7.063 (0.22), 7.301 (0.08), 7.309 (0.09), 7.329 (0.18), 7.337 (0.17), 7.345 (0.17), 7.365 (0.10), 7.373 (0.09).
Beispiel 150A
(+/-)-ie/f-Butyl-5-amino-4-(2,3,6-trichlorphenyl)pentanoat (Racemat)
Eine Lösung von (+/-)-ie/f-Butyl-4-cyan-4-(2,3,6-trichlorphenyl)butanoat (10.2 g, 95% Reinheit, 27.9 mmol, Beispiel 72A) in ie f-Butanol (210 ml) und Methanol (9.1 ml) wurde mit Raney-Nickel (1.64 g, 27.9 mmol) versetzt, und das Gemisch wurde über Nacht unter Rühren bei Normaldruck hydriert. Eine zusätzliche Menge Raney-Nickel (2.0 g, 34.0 mmol) wurde hinzugefügt, und das Gemisch wurde drei weitere Tage unter Rühren bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert, dreimal mit Methanol (jeweils 30 ml) nachgewaschen und das Filtrat eingeengt. Der Rückstand wurde in Ethylacetat (400 ml) aufgenommen. Diese organische Phase wurde zweimal mit 1 M Salzsäure (300 ml) extrahiert. Die vereinigten wäßrigen Phasen wurden durch langsame Zugabe von Natriumhydrogencarbonat auf pH 8-9 ge- bracht und dann zweimal mit Ethylacetat (jeweils 300 ml) extrahiert. Die vereinten organischen Phasen wurden über Natriumsulfat getrocknet und am Rotationsverdampfer eingeengt. Nach Trocknen im Vakuum wurden 2.54 g (89% Reinheit, 23% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.30 min; MS (ESIpos): m/z = 352 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.336 (16.00), 1.352 (1.85), 1.492 (0.42), 2.013 (0.65), 2.022 (0.61 ), 2.030 (0.63), 2.042 (0.89), 2.055 (0.81 ), 2.070 (0.42), 2.082 (0.42), 2.097 (0.43), 3.049 (0.54), 3.069 (0.57), 7.426 (0.52), 7.506 (0.64), 7.532 (0.56), 7.544 (0.61 ), 7.553 (0.41 ).
Beispiel 151A
(+/-)-ie/f-Butyl-6-amino-5-(2-chlorphenyl)hexanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-5-(2-chlorphenyl)-5-cyanpentanoat (2.70 g, 9.19 mmol, Beispiel 73A) in ie/f-Butanol (54 ml) wurde mit Raney-Nickel (540 mg, 9.19 mmol) versetzt und 24 h bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert, zweimal mit ie f-Butanol (15 ml) nachgewaschen und das Filtrat eingeengt. Der Rückstand wurde in Ethylacetat (60 ml) aufgenommen und nacheinander mit 1 M Salzsäure und Wasser extrahiert. Die vereinigten wässrigen Phasen wurden mit gesättigter Natriumhydrogencarbonat-Lösung auf pH 8-9 eingestellt und zweimal mit Ethylacetat (jeweils 80 ml) extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 1.32 g (95% Reinheit, 46% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.33 min; MS (ESIpos): m/z = 298 [M+H]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.1 1 1 (0.02), 1.195 (0.08), 1.271 (0.23), 1.290 (0.45), 1.315 (0.77), 1.334 (1.17), 1.356 (16.00), 1.462 (0.15), 1.477 (0.17), 1.485 (0.23), 1.496 (0.24), 1.509 (0.29), 1.519 (0.27), 1.532 (0.20), 1.542 (0.12), 1.556 (0.09), 1.697 (0.17), 1.710 (0.26), 1.724 (0.27), 1.736 (0.26), 1.744 (0.21 ), 1.769 (0.14), 1.784 (0.08), 1.876 (0.06), 2.074 (0.13), 2.093 (0.23), 2.1 13 (0.58), 2.131 (0.90), 2.144 (0.77), 2.162 (0.39), 2.183 (0.14), 2.201 (0.09), 2.367 (0.06), 2.739 (1.29), 2.756 (1.29), 3.1 10 (0.18), 3.127 (0.32), 3.140 (0.35), 3.150 (0.32), 3.163 (0.27), 3.180 (0.13), 7.171 (0.12), 7.196 (0.28), 7.206 (0.32), 7.21 1 (0.31 ), 7.217 (0.44), 7.227 (0.35), 7.238 (0.29), 7.248 (0.07), 7.293 (0.22), 7.315 (1.26), 7.326 (1.32), 7.405 (0.67), 7.425 (0.53).
Beispiel 152A
(+/-)-ie/f-Butyl-6-amino-5-(2-chlorphenyl)-5-methylhexanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-5-(2-chlorphenyl)-5-cyanhexanoat (2.38 g, 7.73 mmol, Bei- spiel 74A) in ierf-Butanol (45 ml) wurde mit Raney-Nickel (454 mg, 7.73 mmol) versetzt und 24 h bei Normaldruck hydriert. Anschließend wurde erneut mit Raney-Nickel (454 mg, 7.73 mmol) versetzt und weitere 24 h bei Normaldruck hydriert. Anschließend wurde ein weiteres Mal mit Raney- Nickel (454 mg, 7.73 mmol) versetzt und weitere 24 h bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert, zweimal mit ierf-Butanol (15 ml) nachgewaschen und das Filtrat eingeengt. Der Rückstand wurde in Ethylacetat (80 ml) aufgenommen und nacheinander mit 1 M Salzsäure und Wasser (jeweils 80 ml) extrahiert. Die vereinigten wässrigen Pha-
sen wurden mit gesättigter Natriumhydrogencarbonat-Lösung auf pH 8-9 eingestellt und zweimal mit Ethylacetat (jeweils 100 ml) extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 820 mg (61 % Reinheit, 21% d. Th.) der Titelverbindung im Gemisch mit dem de- chlorierten Produkt ((+/-)-ie/f-Butyl-6-amino-5-methyl-5-phenylhexanoat) erhalten.
LC-MS (Methode 1 ): Rt = 1.24 min; MS (ESIpos): m/z = 312 [M+H]+ Beispiel 153A
(+/-)-Methyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}hydratropaat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (257 mg, 750 μηηοΙ, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (4 ml) wurden HATU (428 mg, 1.13 mmol) und DIPEA (390 μΙ, 2.3 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-Methyl-3-amino-2-phenylpropanoat-Hydrochlorid (178 mg, 825 μηηοΙ, Beispiel 75A) hinzugegeben, und das Gemisch wurde 1.5 h bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit 10%-iger Zitronensäure (50 ml) versetzt, und der gebildete Niederschlag wurde abfiltriert, zweimal mit Wasser (5 ml) gewaschen und im Vakuum getrocknet. Anschließend wurde der Niederschlag in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (50 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohe- xan/Ethylacetat 97:3 -> 8:2, Isolera One) gereinigt. Es wurden 175 mg (90% Reinheit, 42% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.16 min; MS (ESIpos): m/z = 547/549 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.64), 0.008 (1.20), 2.167 (2.67), 3.287 (1.39), 3.647 (16.00), 4.106 (0.90), 4.126 (1.46), 7.325 (1.12), 7.336 (1.15), 7.347 (0.78), 7.387 (12.53), 7.398 (7.56), 7.493 (0.93), 7.507 (2.84), 7.514 (2.14), 7.526 (5.17), 7.537 (5.83), 7.543 (3.52), 7.555 (1.01 ), 7.851 (1.24), 7.856 (1.07), 7.873 (1.92), 7.879 (1.75), 7.945 (3.66), 7.968 (2.29), 9.024 (1.33).
Beispiel 154A
(+/-)-Ethyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]am
propanoat (Racemat)
Zu einer Lösung aus (+/-)-Ethyl-3-amino-2-(2-chlorphenyl)propanoat (1.25 g, 5.50 mmol, Beispiel 76A) in Dichlormethan (25 ml) wurden nacheinander Triethylamin (1.4 ml, 10 mmol) und ein Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonylchlorid (1.80 g, 5.00 mmol, Beispiel 1A) in Dichlormethan (25 ml) gegeben, und das Reaktionsgemisch wurde 2 h bei RT gerührt. Anschließend wurde das Gemisch mit Dichlormethan (50 ml) und Wasser (100 ml) versetzt und geschüt- telt. Die wässrige Phase wurde zweimal mit Dichlormethan (jeweils 100 ml) extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (200 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat 9:1 ) gereinigt. Es wurden 2.36 g (100% Reinheit, 86% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.37 min; MS (ESIpos): m/z = 551/553 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.26), 0.008 (1.20), 0.890 (2.24), 1.131 (7.34), 1.149 (16.00), 1.157 (1.41 ), 1.166 (7.62), 1.175 (1.72), 1.193 (0.77), 1.398 (7.12), 1.988 (2.67), 2.184 (1 1.70), 4.021 (1.07), 4.039 (1.23), 4.1 11 (1.87), 4.128 (5.28), 4.146 (5.04), 4.164 (1.63), 4.558 (1.60), 4.577 (3.41 ), 4.596 (1 .41 ), 7.354 (1.78), 7.358 (1.84), 7.372 (1.87), 7.378 (2.92), 7.382 (1.97), 7.397 (2.15), 7.401 (2.27), 7.416 (0.98), 7.419 (0.83), 7.492 (1.87), 7.496 (3.72), 7.502 (3.99), 7.508 (6.94), 7.513 (7.28), 7.519 (4.27), 7.525 (7.98), 7.527 (9.43), 7.531 (6.54), 7.535 (8.60), 7.538 (8.48), 7.542 (5.71 ), 7.545 (4.27), 7.555 (1.57), 7.849 (2.18), 7.854 (1.90), 7.871 (3.29), 7.876 (3.13), 7.944 (5.62), 7.966 (3.53), 9.014 (1.17), 9.029 (2.36), 9.043 (1.14).
Beispiel 155A
(+/-)-Methyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]^
propanoat (Racemat)

Zu einer Suspension aus (+/-)-Methyl-3-amino-2-(2-methoxyphenyl)propanoat-Hydrochlorid (1.52 g, 89% Reinheit, 5.50 mmol, Beispiel 77A) in Dichlormethan (15 ml) wurden nacheinander Triethylamin (1.4 ml, 10 mmol) und eine Suspension aus 6-Brom-3-methyl-2-phenylchinolin-4- carbonylchlorid (902 mg, 2.50 mmol, Beispiel 1A) in Dichlormethan (10 ml) gegeben, und das Gemisch wurde 24 h bei RT gerührt. Anschließend wurde das Gemisch mit Dichlormethan und Wasser (jeweils 100 ml) versetzt und geschüttelt. Die wässrige Phase wurde einmal mit Dichlormethan (100 ml) extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (200 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyc- lohexan/Ethylacetat 8:2) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 1.14 g (100% Reinheit, 85% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.14 min; MS (ESIpos): m/z = 533/535 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.988 (0.57), 2.194 (6.26), 3.303 (2.46), 3.324 (1.41 ), 3.622 (14.45), 3.799 (16.00), 4.364 (0.82), 4.383 (1.55), 4.402 (0.71 ), 6.945 (0.84), 6.947 (0.81 ), 6.964 (1.83), 6.966 (1.71 ), 6.983 (1.08), 7.034 (1.46), 7.053 (1.84), 7.279 (3.27), 7.297 (3.66), 7.301 (1.80), 7.315 (0.75), 7.493 (0.95), 7.504 (1.39), 7.508 (2.60), 7.514 (2.02), 7.521 (1.24), 7.527 (4.31 ), 7.539 (5.20), 7.541 (5.04), 7.546 (3.03), 7.549 (1.98), 7.558 (0.93), 7.850 (1.24), 7.855 (1.01 ), 7.872 (1.86), 7.878 (1.67), 7.943 (3.32), 7.966 (2.06), 8.929 (1.40).
Beispiel 156A
(+/-)-Methyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]a
propanoat (Racemat)

Zu einer Suspension aus (+/-)-Methyl-3-amino-2-(4-fluorphenyl)propanoat-Hydrochlorid (129 mg, 550 μηηοΙ, Beispiel 78A) in Dichlormethan (3 ml) wurden nacheinander Triethylamin (140 μΙ, 1.0 mmol) und eine Suspension aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonylchlorid (180 mg, 500 μηηοΙ, Beispiel 1A) in Dichlormethan (2 ml) gegeben, und das Gemisch wurde 18 h bei RT gerührt. Anschließend wurden erneut (+/-)-Methyl-3-amino-2-(4-fluorphenyl)propanoat- Hydrochlorid (129 mg, 550 μηιοΙ, Beispiel 78A) in Dichlormethan (2 ml) und Triethylamin (140 μΙ, 1.0 mmol) hinzugegeben, und das Reaktionsgemisch wurde weitere 4 Tage bei RT gerührt. Anschließend wurde das Gemisch mit Dichlormethan und Wasser (jeweils 50 ml) versetzt und geschüttelt. Die wässrige Phase wurde einmal mit Dichlormethan (50 ml) extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (50 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat 8:2) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 213 mg (90% Reinheit, 74% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.19 min; MS (ESIpos): m/z = 521/523 [M+H]+
1H-NMR (400 MHz, DMSO-d6): d [ppm] = 9.01 (t, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.71 (br. s, 1 H), 7.57-7.49 (m, 5H), 7.47-7.38 (m, 2H), 7.25-7.18 (m, 2H), 4.14 (t, 1 H), 4.02-3.78 (br. m, 2H), 3.65 (s, 3H), 2.18 (br. s, 3H).
Beispiel 157A
(+/-)-Ethyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]am
(Racemat)

Zu einer Suspension aus (+/-)-Ethyl-3-amino-2-methyl-2-phenylpropanoat-Hydrochlorid (146 mg, 600 mol, darstellbar gemäß Archiv der Pharmazie 1985, 318 (7), 593-600) in Dichlormethan (3 ml) wurden nacheinander Λ/,/V-Diisopropylethylamin (260 μΙ, 1.5 mmol) und eine Suspension aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonylchlorid (180 mg, 500 mol, Beispiel 1A) in Dichlormethan (2 ml) gegeben, und das Gemisch wurde 16 h bei RT gerührt. Anschließend wurde das Gemisch mit Dichlormethan und Wasser (jeweils 30 ml) versetzt und geschüttelt. Die wässrige Phase wurde einmal mit Dichlormethan (30 ml) extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (50 g Kieselgel Biotage Snap-Cartridge KP-Sil, CyclohexanEthylacetat 93:7— > 7:3, Isolera One) gereinigt. Die vereinig- ten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 225 mg (100% Reinheit, 85 % d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.39 min; MS (ESIpos): m/z = 531/533 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.890 (0.83), 1.123 (6.66), 1.141 (13.96), 1.158 (7.14), 1.175 (1.38), 1.192 (0.72), 1.397 (2.24), 1.646 (16.00), 1.988 (2.56), 2.156 (2.64), 3.980 (0.77), 4.020 (0.70), 4.038 (0.69), 4.068 (0.78), 4.077 (0.77), 4.087 (1.02), 4.095 (2.44), 4.105 (1.73), 4.1 13 (2.67), 4.124 (2.66), 4.142 (2.42), 4.150 (1.36), 4.168 (0.87), 7.303 (1.56), 7.318 (1.50), 7.366 (1.45), 7.387 (5.68), 7.399 (7.92), 7.403 (13.69), 7.419 (1.37), 7.480 (0.44), 7.492 (1.60), 7.507 (4.84), 7.512 (3.61 ), 7.525 (6.69), 7.541 (8.34), 7.560 (1.72), 7.720 (0.61 ), 7.841 (1.93), 7.846 (1.67), 7.864 (2.87), 7.869 (2.68), 7.941 (5.55), 7.963 (3.54), 8.721 (1.12), 8.735 (2.00), 8.751 (1.09).
Beispiel 158A
(+/-)-Methyl-2-({[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbony^
butanoat (Racemat)

Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (500 mg, 1.46 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (5 ml) wurden HATU (833 mg, 2.19 mmol) und DIPEA (760 μΙ, 4.4 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-Methyl-2-(aminomethyl)-2-phenylbutanoat-Hydrochlorid (562 mg, 95% Reinheit, 2.19 mmol, darstellbar gemäß Archiv der Pharmazie 1985, 318 {7), 593-600), gelöst in DMF (3 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethylacetat und Wasser (jeweils 20 ml) versetzt und geschüttelt. Die wässrige Phase wurde zweimal mit Ethylacetat (jeweils 20 ml) extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat 9:1 ) gereinigt. Es wurden 567 mg (98% Reinheit, 72% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.24 min; MS (ESIpos): m/z = 531/533 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.63), 0.008 (0.56), 0.793 (2.77), 0.81 1 (6.44), 0.830 (2.94), 1.398 (0.51 ), 2.109 (1.08), 2.127 (3.1 1 ), 2.146 (4.00), 2.163 (2.40), 3.625 (16.00), 4.039 (0.43), 4.052 (0.47), 4.074 (0.65), 4.085 (0.60), 4.202 (0.58), 4.219 (0.62), 4.235 (0.46), 7.271 (0.56), 7.288 (1.55), 7.306 (1.12), 7.325 (2.20), 7.342 (5.33), 7.361 (3.46), 7.379 (2.92), 7.399 (0.99), 7.492 (1.16), 7.507 (3.59), 7.512 (2.64), 7.525 (4.86), 7.540 (5.95), 7.560 (1.26), 7.842 (1.41 ), 7.847 (1.26), 7.864 (2.14), 7.869 (2.02), 7.939 (4.01 ), 7.962 (2.55), 8.639 (0.83), 8.654 (1.55), 8.669 (0.80).
Beispiel 159A
(+/-)-Ethyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]am
phenyl]propanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (52 mg, 153 μηηοΙ, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (1.5 ml) wurden HATU (87 mg, 230 μηηοΙ) und DIPEA (80 μΙ, 460 μηηοΙ) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-Ethyl-3-amino-2-[3-(trifluormethyl)phenyl]propanoat (60 mg, 230 μηηοΙ, Beispiel 80A), gelöst in DMF (1.5 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mittels präparativer HPLC (Methode 22) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und im Vakuum getrocknet. Es wurden 43 mg (98% Reinheit, 47% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.41 min; MS (ESIpos): m/z = 585/587 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.137 (5.86), 1.154 (1 1.79), 1.172 (5.88), 1.195 (3.38), 1.212 (3.46), 1.426 (1 1.61 ), 1.459 (1.63), 2.011 (3.91 ), 2.1 1 1 (2.10), 2.189 (0.44), 2.321 (0.63), 3.656 (0.73), 3.721 (1.10), 3.736 (1.02), 3.898 (0.66), 3.918 (0.84), 3.932 (1.58), 3.952 (2.40), 3.968 (2.49), 3.986 (1.38), 4.099 (0.63), 4.109 (1.09), 4.1 17 (1.61 ), 4.126 (2.89), 4.135 (3.04), 4.144 (3.09), 4.153 (2.68), 4.161 (1.51 ), 4.170 (0.92), 4.179 (0.53), 4.223 (1.36), 4.242 (2.35), 4.261 (1.04), 7.506 (3.76), 7.523 (16.00), 7.565 (0.63), 7.577 (0.56), 7.585 (0.56), 7.597 (0.53), 7.625 (1.00), 7.644 (2.61 ), 7.664 (2.44), 7.699 (3.02), 7.741 (5.52), 7.847 (1.72), 7.852 (1.65), 7.870 (2.67), 7.874 (2.58), 7.941 (4.43), 7.963 (2.81 ), 8.106 (0.43), 8.124 (0.41 ), 8.617 (0.41 ), 8.997 (1.32), 9.010 (2.41 ), 9.023 (1.25).
Beispiel 160A
(+/-)-Ethyl-2-(1 ,3-benzodioxol-5-yl)-3-{[(6-brom-3-methyl-2-phenylchinoli
propanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (144 mg, 421 μηηοΙ, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (5 ml) wurden HATU (240 mg, 632 μηηοΙ) und DIPEA (220 μΙ, 1.3 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-Ethyl-3-amino-2-(1 ,3-benzodioxol-5-yl)propanoat (150 mg, 632 μηηοΙ, Beispiel 81A), gelöst in DMF (4 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mittels präparativer HPLC (Methode 22) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und im Vakuum getrocknet. Es wurden 126 mg (98% Reinheit, 52% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.22 min; MS (ESIpos): m/z = 561/563 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.132 (7.84), 1.150 (16.00), 1.168 (7.82), 2.203 (8.18), 3.788 (0.91 ), 3.889 (0.95), 3.993 (2.51 ), 4.012 (4.07), 4.032 (1.55), 4.043 (0.47), 4.061 (1.04), 4.071 (1.16), 4.079 (1.28), 4.088 (3.86), 4.107 (5.28), 4.125 (3.40), 4.135 (1.13), 4.143 (0.99), 4.153 (0.87), 6.013 (15.21 ), 6.836 (2.25), 6.839 (2.39), 6.855 (4.06), 6.859 (4.32), 6.900 (6.60), 6.920 (3.60), 6.946 (6.37), 6.949 (6.20), 7.485 (0.65), 7.498 (1.98), 7.513 (5.89), 7.518 (4.93), 7.532 (9.29), 7.544 (12.04), 7.562 (2.49), 7.748 (0.84), 7.852 (2.40), 7.857 (2.28), 7.874 (3.60), 7.879 (3.55), 7.948 (6.49), 7.970 (4.10), 8.979 (1.64), 8.992 (3.19), 9.006 (1.64).
Beispiel 161A
(+/-)-fe/f-Butyl-3-{[(6-brom-3-methyl-2-phenylcN
2-methylpropanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (215 mg, 628 μηηοΙ, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (2 ml) wurden HATU (358 mg, 942 μηηοΙ) und DIPEA (330 μΙ, 1.9 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-3-amino-2-(3-chlorpyridin-2-yl)-2-methylpropanoat (255 mg, 942 μηηοΙ, Beispiel 82A), gelöst in DMF (2 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mittels praparativer HPLC (Methode 15) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und im Vakuum getrocknet. Es wurden 230 mg (98% Reinheit, 60% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.36 min; MS (ESIpos): m/z = 594/596 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.405 (16.00), 1.677 (4.42), 2.174 (1.39), 7.369 (0.45), 7.380 (0.48), 7.388 (0.50), 7.400 (0.48), 7.504 (1.05), 7.51 1 (0.83), 7.522 (2.01 ), 7.531 (2.12), 7.839 (0.57), 7.844 (0.42), 7.861 (0.83), 7.866 (0.71 ), 7.906 (0.70), 7.909 (0.69), 7.926 (2.00), 7.948 (0.81 ), 8.519 (0.64), 8.521 (0.63), 8.530 (0.63), 8.671 (0.59).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.67 (t, 1 H), 8.53 (d, 1 H), 7.98-7.89 (m, 2H), 7.85 (dd, 1 H), 7.81 (br. s, 1 H), 7.57-7.46 (m, 5H), 7.38 (dd, 1 H), 4.48 (br. dd, 1 H), 3.92 (br. d, 1 H), 2.17 (br. s, 3H), 1.68 (s, 3H), 1.40 (s, 9H).
Beispiel 162A
(+/-)-Methyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]am
methylpropanoat (Racemat)

Zu einer Suspension aus (+/-)-Methyl-3-amino-2-(2-fluorphenyl)-2-methylpropanoat (127 mg, 600 mol, CAS-RN 1803580-97-3, kommerziell verfügbar) in Dichlormethan (3 ml) wurden nacheinander Λ/,/V-Diisopropylethylamin (260 μΙ, 1.5 mmol) und eine Suspension aus 6-Brom-3- methyl-2-phenylchinolin-4-carbonylchlorid (180 mg, 500 μηηοΙ, Beispiel 1A) in Dichlormethan (2 ml) gegeben, und das Gemisch wurde 16 h bei RT gerührt. Anschließend wurde das Gemisch mit Dichlormethan und Wasser (jeweils 30 ml) versetzt und geschüttelt. Die wässrige Phase wurde einmal mit Dichlormethan (30 ml) extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (50 g Kieselgel Biotage Snap-Cartridge KP-Sil, CyclohexanEthylacetat 93:7— > 7:3, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 208 mg (100% Reinheit, 78% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.25 min; MS (ESIpos): m/z = 535/537 [M+H]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.157 (0.91 ), 1.174 (1.83), 1.192 (0.94), 1.397 (0.91 ), 1.614 (8.25), 1.988 (3.41 ), 2.149 (0.77), 3.31 1 (16.00), 3.931 (0.42), 4.020 (0.84), 4.038 (0.83), 4.199 (0.51 ), 4.216 (0.55), 4.231 (0.46), 4.250 (0.42), 7.171 (0.61 ), 7.192 (0.83), 7.201 (0.75), 7.209 (0.64), 7.222 (1.09), 7.227 (1.32), 7.247 (0.77), 7.371 (0.64), 7.387 (0.59), 7.447 (0.69), 7.467 (1.23), 7.492 (1.21 ), 7.507 (2.39), 7.513 (1.85), 7.525 (3.88), 7.538 (4.70), 7.556 (0.86), 7.840 (0.91 ), 7.845 (0.86), 7.863 (1.41 ), 7.868 (1.37), 7.934 (2.68), 7.956 (1.66), 8.762 (0.58), 8.778 (1.03), 8.793 (0.57).
Beispiel 163A
(+/-)-fe/f-Butyl-3-{[(6-brom-3-methyl-2-phenylcN
2-methylpropanoat (Racemat)

Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (550 mg, 1.61 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (5 ml) wurden HATU (917 mg, 2.41 mmol) und DIPEA (840 μΙ, 4.8 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-3-amino-2-(2,6-difluorphenyl)-2-methylpropanoat (689 mg, 95% Reinheit, 2.41 mmol, Beispiel 83A), gelöst in DMF (3.5 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethylacetat und Wasser (jeweils 20 ml) versetzt und geschüttelt. Die wässrige Phase wurde zweimal mit Ethylacetat (jeweils 20 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan- /Ethylacetat 85:15) gereinigt. Es wurden 645 mg (98% Reinheit, 66% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.59 min; MS (ESIpos): m/z = 595/597 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.401 (16.00), 1.744 (1.68), 7.072 (0.64), 7.096 (0.43), 7.506 (1.02), 7.512 (0.83), 7.524 (1.97), 7.533 (1.98), 7.838 (0.43), 7.860 (0.67), 7.865 (0.64), 7.930 (1.26), 7.953 (0.79), 8.791 (0.50).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.79 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.80-7.47 (m, 6H), 7.45-7.34 (m, 1 H), 7.07 (br. t, 2H), 4.18 (br. dd, 1 H), 3.78 (br. d, 1 H), 2.16 (br. s, 3H), 1.74 (br. s, 3H), 1.40 (s, 9H).
Beispiel 164A
(+/-)-Methyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]^
methylpropanoat (Racemat)

Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (500 mg, 1.46 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (5 ml) wurden HATU (833 mg, 2.19 mmol) und DIPEA (760 μΙ, 4.4 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-Methyl-3-amino-2-(2-chlorphenyl)-2-methylpropanoat (525 mg, 95% Reinheit, 2.19 mmol, verfügbar bei Santai Labs), gelöst in DMF (3 ml) hinzugege- ben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethylacetat und Wasser (jeweils 20 ml) versetzt und geschüttelt. Die wässrige Phase wurde zweimal mit Ethylacetat (jeweils 20 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyc- lohexan/Ethylacetat 85:15) gereinigt. Es wurden 554 mg (98% Reinheit, 67% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.30 min; MS (ESIpos): m/z = 551/553 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.397 (1.34), 1.662 (9.58), 1.988 (0.59), 2.072 (0.62), 2.152 (0.72), 3.640 (16.00), 3.930 (0.48), 3.953 (0.55), 4.405 (0.60), 4.423 (0.64), 4.438 (0.58), 4.457 (0.52), 7.344 (0.97), 7.362 (1.87), 7.377 (1.41 ), 7.398 (0.76), 7.453 (1.58), 7.472 (1.30), 7.491 (1.10), 7.506 (3.18), 7.512 (2.71 ), 7.524 (6.80), 7.536 (6.85), 7.838 (1.13), 7.843 (1.1 1 ), 7.861 (1.78), 7.866 (1.77), 7.930 (3.34), 7.952 (2.02), 8.712 (0.72), 8.729 (1.22), 8.744 (0.69).
Beispiel 165A
(+/-)-Ethyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]a
(Racemat)

Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (171 mg, 500 μηηοΙ, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (5 ml) wurden HATU (150 μΙ, 750 μηηοΙ) und DIPEA (260 μΙ, 1.5 mmol) gegeben und das Gemisch wurde 5 min bei RT gerührt. Anschließend wurde (+/-)-Ethyl-3-amino-2-fluor-2-phenylpropanoat-Hydrochlorid (186 mg, 750 μηηοΙ, CAS-RN 1909308-68-4, kommerziell verfügbar) hinzugegeben, und das Gemisch wurde 2.5 h bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethyl- acetat und Wasser (jeweils 50 ml) versetzt und geschüttelt. Die wässrige Phase wurde einmal mit Ethylacetat (50 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung (80 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash- Säulenchromatographie (50 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat 93:7— > 7:3) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 222 mg (100% Reinheit, 83% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.20 min; MS (ESIpos): m/z = 535/537 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.50), 0.008 (1.61 ), 1.143 (0.70), 1.179 (7.74), 1.197 (16.00), 1.215 (8.00), 1.233 (0.88), 1.397 (1.1 1 ), 2.168 (1.34), 2.327 (0.57), 4.217 (3.02), 4.233 (3.04), 5.754 (1.29), 7.387 (0.85), 7.398 (0.54), 7.478 (5.24), 7.494 (7.07), 7.509 (9.52), 7.514 (8.51 ), 7.528 (12.06), 7.542 (13.27), 7.581 (9.10), 7.599 (6.45), 7.850 (2.41 ), 7.871 (3.52), 7.944 (7.12), 7.966 (4.46), 9.179 (1.98), 9.194 (3.94), 9.209 (1.91 ).
Beispiel 166A
(+/-)-Ethyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]am
methyl)phenyl]propanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (506 mg, 1.48 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (6 ml) wurden HATU (843 mg, 2.22 mmol) und DIPEA (770 μΙ, 4.4 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-Ethyl-3-amino-2-methyl-2-[3-(trifluormethyl)phenyl]- propanoat (610 mg, 2.22 mmol, Beispiel 84A), gelöst in DMF (3 ml) hinzugegeben, und das Ge- misch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mittels präparativer HPLC (Methode 15) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 577 mg (98% Reinheit, 64% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.50 min; MS (ESIpos): m/z = 599/601 [M+H]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.124 (6.68), 1.142 (14.17), 1.160 (6.91 ), 1.398 (0.94), 1.694 (16.00), 2.074 (1.16), 3.939 (1.13), 3.952 (1.23), 3.973 (1.51 ), 3.986 (1.41 ), 4.100 (0.67), 4.109 (0.82), 4.1 18 (0.87), 4.127 (3.00), 4.144 (4.39), 4.162 (2.88), 4.170 (0.82), 4.179 (0.79), 4.188 (0.64), 4.219 (1.62), 4.237 (1.71 ), 4.253 (1.39), 4.271 (1.30), 7.478 (0.48), 7.493 (1.25), 7.503 (4.16), 7.510 (3.72), 7.521 (10.32), 7.525 (9.36), 7.621 (0.97), 7.641 (2.45), 7.660 (2.39), 7.686 (2.87), 7.706 (5.92), 7.732 (2.76), 7.751 (1.95), 7.837 (1.69), 7.842 (1.63), 7.859 (2.60), 7.864 (2.57), 7.935 (5.22), 7.957 (3.35), 8.766 (1.12), 8.782 (1.97), 8.796 (1.16).
Beispiel 167A
(+/-)-ie f-Butyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]a
propanoat (Racemat)

Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (681 mg, 1.99 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (6.8 ml) wurden HATU (1.14 g, 2.99 mmol) und DIPEA (1.4 ml, 8.0 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-3-amino-2-(3-methoxyphenyl)propanoat (750 mg, 2.99 mmol, Beispiel 85A), gelöst in DMF (4.6 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Wasser (60 ml) versetzt und geschüttelt. Die wässrige Phase wurde zweimal mit Ethylacetat (100 ml und 80 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde mittels Flash-Säulenchromatographie (50 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyc- lohexan/Ethylacetat 100:0 -> 7:3, Isolera) gereinigt. Es wurden 940 mg (92% Reinheit, 76% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.28 min; MS (ESIpos): m/z = 575/577 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.84), 0.008 (0.62), 1.375 (16.00), 1.398 (7.07), 2.184 (1.00), 2.523 (0.41 ), 3.749 (6.57), 3.960 (0.51 ), 6.879 (0.43), 6.898 (1.61 ), 6.923 (0.54), 6.943 (0.62), 7.304 (0.59), 7.509 (0.84), 7.516 (0.75), 7.529 (1 .92), 7.535 (1.86), 7.542 (1.36), 7.849 (0.46), 7.871 (0.69), 7.876 (0.65), 7.945 (1.19), 7.967 (0.76), 8.992 (0.54).
Beispiel 168A
(+/-)-fe/f-Butyl-3-{[(6-brom-3-methyl-2-phenylcN
propanoat (Racemat)

Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (681 mg, 1.99 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (4.5 ml) wurden HATU (743 mg, 1.95 mmol) und DIPEA (910 μΙ, 5.2 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-3-amino-2-(3-chlorphenyl)propanoat (500 mg, 1.95 mmol, Beispiel 86A), gelöst in DMF (3.0 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Wasser (40 ml) versetzt und geschüttelt. Die wässrige Phase wurde zweimal mit Ethylacetat (70 ml und 50 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde mittels Flash-Säulenchromatographie (50 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan- /Ethylacetat 100:0 -> 7:3, Isolera) gereinigt. Es wurden 430 mg (88% Reinheit, 50% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.36 min; MS (ESIpos): m/z = 579/581 [M+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.98 (t, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.68 (br. s, 1 H), 7.56-7.47 (m, 5H), 7.46-7.32 (m, 4H), 4.01 (t, 1 H), 3.93-3.76 (m, 2H), 2.15 (br. s, 3H), 1.38 (s, 9H).
Beispiel 169A
(+/-)-fe/f-Butyl-3-{[(6-brom-3-methyl-2-phenylcN
propanoat (Racemat)

Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (678 mg, 1.98 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (6.8 ml) wurden HATU (1.13 g, 2.97 mmol) und DIPEA (1.4 ml, 7.9 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-3-amino-2-(3-methylphenyl)propanoat (699 mg, 2.97 mmol, Beispiel 87A), gelöst in DMF (4.6 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Wasser (60 ml) versetzt und geschüttelt. Die wässrige Phase wurde zweimal mit Ethylacetat (1 10 ml und 80 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid- Lösung (60 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde mittels Flash-Säulenchromatographie (50 g Kieselgel Biotage Snap-Cartridge KP- Sil, Cyclohexan/Ethylacetat 100:0— > 7:3, Isolera) gereinigt. Es wurden 830 mg (95% Reinheit, 71 % d. Th.) der Titelverbindung erhalten.
C-MS (Methode 1 ): Rt = 2.56 min; MS (ESIpos): m/z = 559/561 [M+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.99 (t, 1 H), 7.95 (d, 1 H), 7.87 (dd, 1 H), 7.76 (br. s, 1 H), 7.59-7.44 (m, 5H), 7.27 (t, 1 H), 7.19-7.07 (m, 3H), 3.99-3.90 (m, 1 H), 3.89-3.80 (m, 1 H), 3.78-3.66 (m, 1 H), 2.31 (s, 3H), 2.19 (br. s, 2H), 1.37 (s, 9H).
Beispiel 170A
(+/-)-Methyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)cato^
methoxyphenyl)propanoat (Racemat)

Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (506 mg, 1.48 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (7.5 ml) wurden HATU (150 μΙ, 2.2 mmol) und DIPEA (770 μΙ, 4.4 mmol) gegeben und das Gemisch wurde 5 min bei RT gerührt. Anschließend wurde (+/-)-Methyl-3-amino-2-hydroxy-2-(2-methoxyphenyl)propanoat (500 mg, 2.22 mmol, verfügbar bei Santai Labs, Art Nr. ADH-GM-15173) hinzugegeben, und das Gemisch wurde 8.5 h bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethylacetat und Wasser (jeweils 100 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase wurde einmal mit Ethylacetat (100 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung (80 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap- Cartridge KP-Sil, Cyclohexan/Ethylacetat 93:7 — > 4:6, Isolera) gereinigt. Es wurden 586 mg (100% Reinheit, 72% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.98 min; MS (ESIpos): m/z = 549/551 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.91 ), 0.008 (0.93), 1.988 (0.67), 2.092 (1.03), 2.522 (0.43), 3.310 (16.00), 3.635 (12.16), 6.293 (3.96), 6.939 (0.57), 6.958 (1.16), 6.977 (0.66), 6.997 (1.00), 7.017 (1.16), 7.280 (0.42), 7.300 (0.67), 7.479 (0.57), 7.482 (0.55), 7.494 (1.81 ), 7.501 (1.68), 7.514 (4.48), 7.518 (4.24), 7.526 (2.88), 7.538 (0.56), 7.590 (0.79), 7.609 (0.74), 7.813 (0.84), 7.818 (0.78), 7.835 (1.31 ), 7.840 (1.30), 7.899 (2.67), 7.921 (1.57), 8.478 (0.46), 8.493 (0.78), 8.508 (0.45).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.49 (t, 1 H), 7.91 (d, 1 H), 7.83 (dd, 1 H), 7.71 (br. s, 1 H), 7.60 (d, 1 H), 7.55-7.46 (m, 5H), 7.30 (t, 1 H), 7.05-6.92 (m, 2H), 6.29 (s, 1 H), 4.35-4.1 1 (m, 1 H), 4.09-3.93 (m, 1 H), 3.76 (s, 3H), 3.64 (s, 3H), 2.09 (br. s, 3H).
Trennunq der Enantiomere:
Die Titelverbindung (309 mg) wurde in einem Gemisch aus Ethanol (2 ml) und Dichlormethan (3 ml) gelöst und mittels praparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 171A und 172A) [Säule: Daicel Chiralpak IA, 5 μηι, 250 mm x 20 mm; Fluss: 15 ml/min; Detektion: 220 nm; Temperatur: 50 °C; Injektion: 0.15 ml; Eluent: 100% Ethanol; Laufzeit 18 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde in Aceton itril/Wasser lyophilisiert.
Beispiel 171A
(-)-Methyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-hydroxy-2-(2-methoxy- phenyl)propanoat (Enantiomer 1)
Bei der in Beispiel 170A beschriebenen Enantiomerentrennung wurden 145 mg (100% Reinheit, ee-Wert 99%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -68.6°, 589 nm, c = 0.34 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.00 min; MS (ESIpos): m/z = 549/551 [M+H]+
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: 2.095 (0.99), 3.465 (0.46), 3.473 (0.45), 3.487 (0.43), 3.636 (16.00), 6.945 (0.70), 6.960 (1.40), 6.975 (0.80), 6.999 (1.22), 7.016 (1.33), 7.285 (0.49), 7.300 (0.76), 7.314 (0.43), 7.474 (0.42), 7.482 (1.02), 7.490 (1.00), 7.494 (1.82), 7.499 (2.36), 7.514 (4.54), 7.522 (4.89), 7.526 (5.82), 7.538 (0.96), 7.596 (0.92), 7.610 (0.88), 7.818 (1.06), 7.822 (1.03), 7.836 (1.53), 7.840 (1.50), 7.903 (3.12), 7.921 (2.06), 8.485 (0.61 ), 8.497 (1.00), 8.508 (0.60).
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: 8.50 (t, 1 H), 7.91 (d, 1 H), 7.83 (dd, 1 H), 7.67 (br. s, 1 H), 7.60 (d, 1 H), 7.56-7.46 (m, 5H), 7.30 (t, 1 H), 7.05-6.92 (m, 2H), 6.29 (br. s, 1 H), 4.22 (br. s, 1 H), 4.01 (br. s, 1 H), 3.76 (s, 3H), 3.64 (s, 3H), 2.09 (br. s, 3H).
Beispiel 172A
(+)-Methyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-hydroxy-2-(2-methoxy- phenyl)propanoat (Enantiomer 2)
Bei der in Beispiel 170A beschriebenen Enantiomerentrennung wurden 137 mg (100% Reinheit, ee-Wert 99%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = +71.8°, 589 nm, c = 0.33 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.00 min; MS (ESIpos): m/z = 549/551 [M+H]+
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: 2.095 (1.37), 3.762 (16.00), 6.946 (0.95), 6.960 (1.90), 6.975 (1.09), 7.000 (1.66), 7.016 (1.81 ), 7.286 (0.67), 7.300 (1.05), 7.315 (0.59), 7.475 (0.56),
7.484 (1.38), 7.492 (1.40), 7.496 (2.52), 7.501 (3.23), 7.515 (6.13), 7.524 (6.70), 7.527 (7.88), 7.539 (1.35), 7.596 (1.28), 7.61 1 (1.22), 7.821 (1.43), 7.824 (1.40), 7.839 (2.07), 7.842 (2.05), 7.905 (4.13), 7.923 (2.71 ), 8.488 (0.85), 8.499 (1.37), 8.51 1 (0.83).
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: 8.50 (t, 1 H), 7.91 (d, 1 H), 7.83 (dd, 1 H), 7.68 (br. s, 1 H), 7.60 (d, 1 H), 7.55-7.44 (m, 5H), 7.30 (t, 1 H), 7.07-6.91 (m, 2H), 6.28 (br. s, 1 H), 4.22 (br. s, 1 H), 4.01 (br. s, 1 H), 3.76 (s, 3H), 3.64 (s, 3H), 2.10 (br. s, 3H).
Beispiel 173A
(+/-)-fe/f-Butyl-3-{[(6-brom-3-methyl-2-phe^
phenyl)propanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (192 mg, 561 μηηοΙ, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (2.0 ml) wurden HATU (256 mg, 673 μηηοΙ) und DIPEA (230 μΙ, 1.3 mmol) gegeben und das Gemisch wurde 15 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-3-amino-2-(2-fluor-6-methoxyphenyl)propanoat (213 mg, 71% Reinheit, 561 μηηοΙ, Beispiel 88A) hinzugegeben, und das Gemisch wurde 2 h bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethylacetat und Wasser (jeweils 50 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase wurde einmal mit Ethylacetat (30 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung (80 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash- Säulenchromatographie (50 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat 93:7— > 7:3, Isolera One) vorgereinigt. Anschließend wurde das vorgereingte Produkt mittels präparativer HPLC (Methode 15) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 192 mg (100% Reinheit, 58% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.30 min; MS (ESIpos): m/z = 593/595 [M+H]+
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: 8.80 (t, 1 H), 7.94 (d, 1 H), 7.86 (dd, 1 H), 7.78 (br. s, 1 H),
7.55- 7.47 (m, 5H), 7.34-7.26 (m, 1 H), 6.88 (d, 1 H), 6.82 (t, 1 H), 4.34 (dd, 1 H), 4.14-4.05 (m, 1 H), 3.80 (s, 3H), 3.71 (br. s, 1 H), 2.18 (s, 3H), 1.37 (s, 9H).
Trennung der Enantiomere:
Die Titelverbindung (224 mg, vereinigt mit einer aus einem Vorversuch stammenden Menge) wurde in Methanol (10 ml) gelöst und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 174A und 175A) [Säule: Chiralpak AD SFC, 5 μηι, 250 mm x 20 mm; Fluss: 60 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 0.50 ml; Eluent: 70% Kohlendioxid / 30% Ethanol; Laufzeit 1 1 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet.
Beispiel 174A
(-)-ie/f-Butyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(2-fluor-6-methoxy- phenyl)propanoat (Enantiomer 1)
Bei der in Beispiel 173A beschriebenen Enantiomerentrennung wurden 81 mg (100% Reinheit, ee-Wert 100%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -62.0°, 589 nm, c = 0.33 g/100 ml, Chloroform
LC-MS (Methode 2): Rt = 1.30 min; MS (ESIpos): m/z = 593/595 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.364 (16.00), 2.181 (2.78), 3.794 (5.53), 6.818 (0.64), 6.869 (0.67), 6.890 (0.73), 7.289 (0.41 ), 7.307 (0.40), 7.502 (0.87), 7.510 (0.77), 7.522 (2.43), 7.526 (2.34), 7.534 (1.34), 7.840 (0.46), 7.846 (0.40), 7.863 (0.71 ), 7.868 (0.66), 7.930 (1.25), 7.952 (0.76), 8.795 (0.59).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.79 (t, 1 H), 7.94 (d, 1 H), 7.86 (dd, 1 H), 7.77 (br. s, 1 H),
7.56- 7.46 (m, 5H), 7.35-7.25 (m, 1 H), 6.88 (d, 1 H), 6.82 (t, 1 H), 4.34 (dd, 1 H), 4.14-4.04 (m, 1 H), 3.79 (s, 3H), 3.75-3.65 (m, 1 H), 2.18 (s, 3H), 1.36 (s, 9H). Beispiel 175A
(+)-ie/f-Butyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(2-fluor-6-methoxy- phenyl)propanoat (Enantiomer 2)
Bei der in Beispiel 173A beschriebenen Enantiomerentrennung wurden 92 mg (100% Reinheit, ee-Wert 100%) der Titelverbindung als später eluierendes Enantiomer erhalten. [a]D 20 = +60.9°, 589 nm, c = 0.32 g/100 ml, Chloroform
LC-MS (Methode 2): Rt = 1.30 min; MS (ESIpos): m/z = 593/595 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.364 (16.00), 2.181 (2.95), 3.795 (5.53), 6.818 (0.67), 6.841 (0.40), 6.870 (0.70), 6.891 (0.77), 7.290 (0.44), 7.308 (0.43), 7.502 (0.93), 7.510 (0.82), 7.523 (2.63), 7.526 (2.55), 7.841 (0.47), 7.846 (0.41 ), 7.863 (0.72), 7.868 (0.67), 7.930 (1.24), 7.953 (0.76), 8.795 (0.63).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.79 (t, 1 H), 7.94 (d, 1 H), 7.86 (dd, 1 H), 7.77 (br. s, 1 H), 7.56-7.46 (m, 5H), 7.35-7.24 (m, 1 H), 6.88 (d, 1 H), 6.82 (t, 1 H), 4.34 (dd, 1 H), 4.16-4.03 (m, 1 H), 3.79 (s, 3H), 3.71 (br. s, 1 H), 2.18 (s, 3H), 1.36 (s, 9H).
Beispiel 176A
(+/-)-fe/f-Butyl-3-{[(6-brom-3-methyL
2-methylpropanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (782 mg, 2.29 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (10 ml) wurden HATU (1.30 g, 3.43 mmol) und DIPEA (1.2 ml, 6.9 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-3-amino-2-(2-methoxyphenyl)-2-methylpropanoat (1.30 g, 70% Reinheit, 3.43 mmol, Beispiel 89A), gelöst in DMF (5 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethylacetat und Wasser (jeweils 50 ml) versetzt. Nach Schütteln und Phasentrennung wurde die wässrige Phase zweimal mit Ethylacetat (jeweils 50 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel, Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat 93:7— > 6:4, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde in Acetonitril/Wasser lyophilisiert. Es wurden 1.09 g (98% Reinheit, 79% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.55 min; MS (ESIpos): m/z = 589/591 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.39 (dd, 1 H), 7.92 (d, 1 H), 7.84 (dd, 1 H), 7.65 (br. s, 1 H), 7.55-7.45 (m, 5H), 7.30-7.20 (m, 2H), 6.98 (d, 1 H), 6.93 (t, 1 H), 4.29 (dd, 1 H), 3.78 (s, 3H), 3.72 (dd, 1 H), 2.08 (br. s, 3H), 1.57 (s, 3H), 1.36 (s, 9H).
Trennung der Enantiomere:
Die Titelverbindung (1.00 g) wurde in Ethanol (25 ml) gelöst und mittels präparativer SFC an chi- raler Phase in die Enantiomere getrennt (siehe Beispiele 177A und 178A) [Säule: Daicel Chiral- pak AD, 20 μηη, 360 mm x 50 mm; Fluss: 140 ml/min; Detektion: 210 nm; Temperatur: 38 °C; Injektion: 2.0 ml; Eluent: 70% Kohlendioxid / 30% Ethanol -> 50% Kohlendioxid / 50% Ethanol; Laufzeit 9 min]. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet.
Beispiel 177A
(-)-ie/f-Butyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(2-methoxyphenyl)-2- methylpropanoat (Enantiomer 1)
Bei der in Beispiel 176A beschriebenen Enantiomerentrennung wurde die vorgereinigte Titelver- bindung als früher eluierendes Enantiomer (ee-Wert 100%) erhalten und mittels präparativer HPLC (Methode 20) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt und lyophilisiert. Es wurden 440 mg (98% Reinheit) der Titelverbindung erhalten.
[a]D 20 = -95.7°, 589 nm, c = 0.35 g/100 ml, Chloroform
LC-MS (Methode 2): Rt = 1.35 min; MS (ESIpos): m/z = 589/591 [M+H]+
1H-NMR (600 MHz, DMSO-d6) δ [ppm]: 8.41 (dd, 1 H), 7.92 (d, 1 H), 7.84 (dd, 1 H), 7.60 (br. s, 1 H), 7.55-7.46 (m, 5H), 7.30-7.20 (m, 2H), 6.98 (d, 1 H), 6.93 (t, 1 H), 4.30 (dd, 1 H), 3.78 (s, 3H), 3.75-3.68 (m, 1 H), 2.08 (br. s, 3H), 1.57 (s, 3H), 1.36 (s, 9H).
Beispiel 178A
(+)-ie/f-Butyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(2-methoxyphenyl)-2- methylpropanoat (Enantiomer 2)
Bei der in Beispiel 176A beschriebenen Enantiomerentrennung wurde die vorgereinigte Titelverbindung als später eluierendes Enantiomer (ee-Wert 100%) erhalten und mittels präparativer HPLC (Methode 20) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt und lyophilisiert. Es wurden 450 mg (98% Reinheit) der Titelverbindung erhalten. [a]D 20 = +93.1 °, 589 nm, c = 0.36 g/100 ml, Chloroform
LC-MS (Methode 2): Rt = 1.35 min; MS (ESIpos): m/z = 589/591 [M+H]+
1H-NMR (600 MHz, DMSO-d6) δ [ppm]: 1.364 (16.00), 1.568 (2.53), 6.988 (0.40), 7.228 (0.68), 7.241 (0.64), 7.498 (0.58), 7.503 (0.80), 7.517 (2.34), 7.524 (1.93), 7.848 (0.52), 7.850 (0.50), 7.917 (0.99), 7.931 (0.69), 8.408 (0.45).
1H-NMR (600 MHz, DMSO-d6) δ [ppm]: 8.41 (dd, 1 H), 7.92 (d, 1 H), 7.84 (dd, 1 H), 7.63 (br. s, 1 H), 7.55-7.46 (m, 5H), 7.31 -7.19 (m, 2H), 6.98 (d, 1 H), 6.93 (t, 1 H), 4.29 (dd, 1 H), 3.78 (s, 3H), 3.75-3.67 (m, 1 H), 2.08 (br. s, 3H), 1.57 (s, 3H), 1.36 (s, 9H).
Beispiel 179A
(+/-)-fe/f-Butyl-3-{[(6-brom-3-methyl-2-phenylcN
phenyl)-2-methylpropanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (694 mg, 2.03 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (15 ml) wurden HATU (1.16 g, 3.04 mmol) und DIPEA (1.1 ml, 6.1 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ierf-Butyl-3-amino-2-(2-chlor-6-fluorphenyl)-2-methylpropanoat (1.25 g, 70% Reinheit, 3.04 mmol, Beispiel 90A), gelöst in DMF (5 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethylacetat und Wasser (jeweils 50 ml) versetzt. Nach Schütteln und Phasentrennung wurde die wässrige Phase zweimal mit Ethylacetat (jeweils 50 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natri- umsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel, Biotage Snap-Cartridge KP- Sil, Cyclohexan/Ethylacetat 93:7— > 6:4, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde in Acetonitril/Wasser lyophilisiert. Es wurden 960 mg (83% Reinheit, 64% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.63 min; MS (ESIpos): m/z = 61 1/613 [M+H]+
LC-MS (Methode 7): Rt = 1.71 min
Trennunq der Enantiomere:
Die Titelverbindung (960 mg) wurde in Methanol (35 ml) gelöst und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 180A und 181A) [Säule: Daicel Chi- ralpak AD, 250 mm x 20 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 0.8 ml; Eluent: 80% Kohlendioxid / 20% Methanol; Laufzeit 7 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet.
Beispiel 180A
(+)-ie/f-Butyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(2-chlor-6-fluor- phenyl)-2-methylpropanoat (Enantiomer 1)
Bei der in Beispiel 179A beschriebenen Enantiomerentrennung wurde die vorgereinigte Titelverbindung als früher eluierendes Enantiomer (ee-Wert >99%) erhalten und zweimal mittels präparativer HPLC (Methode 20) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 205 mg (98% Reinheit, 21 % d. Th.) der Titelverbindung erhalten. [a]D 20 = +65.8°, 589 nm, c = 0.40 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.65 min; MS (ESIpos): m/z = 61 1/613 [M+H]+
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: 8.77 (t, 1 H), 7.94 (d, 1 H), 7.86 (dd, 1 H), 7.68 (br. s, 1 H), 7.58-7.47 (m, 5H), 7.42-7.28 (m, 2H), 7.23-7.13 (m, 1 H), 4.47 (br. s, 1 H), 3.81 (br. s, 1 H), 2.16 (br. s, 3H), 1.84 (d, 3H), 1.42 (s, 9H). Beispiel 181A
(-)-ie/f-Butyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(2-chlor-6-fluor- phenyl)-2-methylpropanoat (Enantiomer 2)
Bei der in Beispiel 179A beschriebenen Enantiomerentrennung wurde die vorgereinigte Titelverbindung als später eluierendes Enantiomer (ee-Wert >99%) erhalten und mittels präparativer HPLC (Methode 20) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 289 mg (98% Reinheit, 30% d. Th.) der Titelverbindung erhalten.
[a]D 20 = -70.6°, 589 nm, c = 0.36 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.67 min; MS (ESIpos): m/z = 61 1/613 [M+H]+
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: 8.76 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.68 (br. s, 1 H), 7.57-7.46 (m, 5H), 7.41 -7.28 (m, 2H), 7.22-7.13 (m, 1 H), 4.47 (br. s, 1 H), 3.80 (br. s, 1 H), 2.16 (br. s, 3H), 1.83 (d, 3H), 1.42 (s, 9H).
Beispiel 182A
(+/-)-fe/f-Butyl-4-{[(6-brom-3-methyl-2-phenylcN
butanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (507 mg, 1.48 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (5 ml) wurden HATU (846 mg, 2.22 mmol) und DIPEA (770 μΙ, 4.4 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend (+/-)-ierf-Butyl-4-amino-3-(2-chlorphenyl)butanoat (600 mg, 2.22 mmol, Beispiel 91A), gelöst in DMF (4 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C ge- rührt. Nach Abkühlen auf RT wurde das Gemisch mittels präparativer HPLC (Methode 22) gereinigt. Es wurden 527 mg (98% Reinheit, 58% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.54 min; MS (ESIpos): m/z = 593/595 [M+H]+
Das Ή-NMR wurde von der Produktcharge (104 mg, 98% Reinheit, 89% d. Th.) eines analog durchgeführten Experiments (ausgehend von 66 mg, 19.3 mmol 6-Brom-3-methyl-2-phenyl- chinolin-4-carbonsäure) erhalten:
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.86 (t, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.78-7.62 (m, 1 H), 7.58-7.47 (m, 6H), 7.44 (dd, 1 H), 7.35 (t, 1 H), 7.27 (t, 1 H), 4.00-3.87 (m, 1 H), 3.84-3.57 (br. m, 2H, teilweise verdeckt), 2.82-2.60 (m, 2H), 2.16 (s, 3H), 1.25 (s, 9H).
Beispiel 183A
(+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(3-chlorphenyl)- butanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (99 mg, 291 μηηοΙ, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (1.5 ml) wurden HATU (166 mg, 436 μηηοΙ) und DIPEA (150 μΙ, 870 μηηοΙ) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-4-amino-3-(3-chlorphenyl)butanoat (294 mg, 40% Reinheit, 436 μηηοΙ, Beispiel 92A), gelöst in DMF (1 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60°C gerührt. Nach Abkühlen auf RT wurde das Gemisch mittels präparativer HPLC (Methode 21 ) gereinigt. Es wurden 224 mg (66% Reinheit, 86% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.53 min; MS (ESIpos): m/z = 591/593 (M+H)+ Beispiel 184A
(+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-methylphenyl)- butanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (629 mg, 1.84 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (6.5 ml) wurden HATU (1.05 g, 2.76 mmol) und DIPEA (960 μΙ, 5.5 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-4-amino-3-(2-methylphenyl)butanoat (724 mg, 95% Reinheit, 2.76 mmol, Beispiel 93A), gelöst in DMF (3.5 ml) hinzugegeben, und das Gemisch
wurde über Nacht bei 60°C gerührt. Nach Abkühlen auf RT wurde das Gemisch mittels praparativer HPLC (Methode 21 ) gereinigt. Es wurden 778 mg (95% Reinheit, 70% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.56 min; MS (ESIpos): m/z = 573/575 (M+H)+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.88 (t, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.69 (br. s, 1 H), 7.59-7.44 (m, 5H), 7.34 (d, 1 H), 7.26-7.03 (m, 3H), 3.80-3.62 (m, 2H), 3.57-3.38 (m, 1 H), 2.78-2.67 (m, 1 H), 2.64-2.56 (m, 1 H), 2.39 (s, 3H), 2.14 (br. s, 2H), 1.22 (s, 9H).
Beispiel 185A
(+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2,6-dichlorphenyl)- butanoat
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (180 mg, 525 μηηοΙ, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (4.5 ml) wurden HATU (300 mg, 788 μηηοΙ) und DIPEA (270 μΙ, 1.6 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ierf-Butyl-4-amino-3-(2,6-dichlorphenyl)butanoat (255 mg, 94% Reinheit, 788 μηηοΙ, Beispiel 94A), gelöst in DMF (2 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60°C gerührt. Nach Abkühlen auf RT wurde das Gemisch mittels praparativer HPLC (Methode 21 ) gereinigt. Es wurden 150 mg (91% Reinheit, 41 % d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.41 min; MS (ESIpos): m/z = 627/629/631 (M+H)+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.96 (t, 1 H), 7.99-7.93 (m, 1 H), 7.90-7.83 (m, 1 H), 7.60-7.38 (m, 8H), 7.36-7.26 (m, 1 H), 4.38 (br. s, 1 H), 4.07 (br. s, 1 H), 3.78 (br. s, 1 H, verdeckt), 3.04 (dd, 1 H), 2.82 (dd, 1 H), 2.19 (br. s, 3H), 1.24 (s, 9H).
Beispiel 186A
(+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(3-methoxyphenyl)- butanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (387 mg, 1.13 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (4.5 ml) wurden HATU (645 mg, 1.70 mmol) und DIPEA (590 μΙ, 3.4 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-4-amino-3-(3-methoxyphenyl)butanoat (500 mg, 90% Reinheit, 1.70 mmol, Beispiel 95A), gelöst in DMF (2 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60°C gerührt. Nach Abkühlen auf RT wurde das Gemisch mittels präpara- tiver HPLC (Methode 21 ) gereinigt. Es wurden 225 mg (93% Reinheit, 31 % d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.42 min; MS (ESIpos): m/z = 589/591 [M+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.81 (t, 1 H), 7.94 (d, 1 H), 7.86 (dd, 1 H), 7.66 (br. s, 1 H), 7.57-7.46 (m, 5H), 7.23 (t, 1 H), 6.92-6.86 (m, 2H), 6.83-6.74 (m, 1 H), 3.80-3.66 (m, 1 H, teilweise verdeckt), 3.72 (s, 3H), 3.66-3.48 (m, 1 H), 3.43-3.23 (m, 1 H), 2.72 (dd, 1 H), 2.59-2.52 (m, 1 H, teilweise verdeckt), 2.1 1 (br. s, 3H), 1.27 (s, 9H). Beispiel 187A
(+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(3-chlorpyridin-2-yl)- 3-methylbutanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (493 mg, 1.44 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (4 ml) wurden HATU (822 mg, 2.16 mmol) und DIPEA (0.75 ml, 4.33 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-4-amino-3-(3-chlorpyridin-2-yl)-3-methylbutanoat (616 mg, 2.16 mmol, Beispiel 96A), gelöst in DMF (4 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60°C gerührt. Nach Abkühlen auf RT wurde das Gemisch mittels praparativer HPLC (Methode 21 ) gereinigt. Es wurden 694 mg (98% Reinheit, 77% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.35 min; MS (ESIpos): m/z = 608/610 [M+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.69 (t, 1 H), 8.48 (dd, 1 H), 7.94 (d, 1 H), 7.86 (dd, 2H), 7.71 (d, 1 H), 7.58-7.44 (m, 5H), 7.32 (dd, 1 H), 4.12 (br. s, 1 H), 3.99 (br. s, 1 H), 3.32 (d, 1 H), 2.67 (d, 1 H), 2.16 (s, 3H), 1.68 (s, 3H), 1.20 (s, 9H)
Beispiel 188A
(+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-chlor-6-fluor- phenyl)butanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (4.95 g, 14.5 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (65 ml) wurden HATU (8.26 g, 21.7 mmol) und DIPEA (7.6 ml, 43 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-4-amino-3-(2-chlor-6-fluorphenyl)butanoat (5.00 g, 17.4 mmol, Beispiel 97A), gelöst in DMF (20 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60°C gerührt. Nach Abkühlen auf RT wurde das Gemisch eingeengt und der Rückstand mit jeweils 200 ml Ethylacetat und Wasser versetzt und geschüttelt. Die wässrige Phase wurde einmal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden einmal mit 200 ml gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulen-
chromatographie (500 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat 9:1— > 8:2) gereinigt. Es wurden 5.23 g (100% Reinheit, 59% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.33 min; MS (ESIpos): m/z = 61 1/613 [M+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.97 (t, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.78-7.59 (br. m, 1 H), 7.60-7.44 (m, 5H), 7.38-7.29 (m, 2H), 7.26-7.12 (m, 1 H), 4.16-3.98 (m, 1 H), 3.93-3.56 (br. m, 2H), 2.85-2.61 (m, 2H), 2.17 (br. s, 3H), 1.24 (s, 9H).
Trennung der Enantiomere:
Die Titelverbindung (5.15 g) wurde in 50 ml Methanol aufgenommen, filtriert und mittels präpara- tiver SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 189A und 190A) [Säu- le: Daicel Chiralcel OJ-H, 5 μηη, 250 mm x 30 mm; Fluss: 175 ml/min; Detektion: 210 nm; Temperatur: 38 °C; Injektion: 0.4 ml; Eluent: 80% Kohlendioxid / 20% Isopropanol; Laufzeit 8 min, isokratisch].
Beispiel 189A
(-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-chlor-6-fluor- phenyl)butanoat (Enantiomer 1 )
Bei der in Beispiel 188A beschriebenen Enantiomerentrennung wurden 1.89 g (100% Reinheit, ee-Wert 97%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -16.6°, 589 nm, c = 0.50 g/100 ml, Chloroform
LC-MS (Methode 1 ): Rt = 2.52 min; MS (ESIpos): m/z = 61 1/613 [M+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.97 (t, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.78-7.57 (m, 1 H), 7.58-7.43 (m, 5H), 7.40-7.28 (m, 2H), 7.26-7.13 (m, 1 H), 4.17-3.99 (m, 1 H), 3.94-3.59 (br. m, 2H), 2.88-2.61 (m, 2H), 2.17 (br. s, 3H), 1.24 (s, 9H).
Beispiel 190A
(+)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-chlor-6-fluor- phenyl)butanoat (Enantiomer 2)
Bei der in Beispiel 188A beschriebenen Enantiomerentrennung wurden 1.85 g (98% Reinheit, ee-Wert 95%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = +16.1 °, 589 nm, c = 0.52 g/100 ml, Chloroform
LC-MS (Methode 1 ): Rt = 2.52 min; MS (ESIpos): m/z = 61 1/613 [M+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.97 (t, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.79-7.60 (br. m, 1 H), 7.56-7.44 (m, 5H), 7.41 -7.27 (m, 2H), 7.26-7.13 (m, 1 H), 4.18-4.00 (m, 1 H), 3.97-3.61 (br. m, 2H), 2.85-2.61 (m, 2H), 2.17 (br. s, 3H), 1.24 (s, 9H).
Beispiel 191A
(+/-)-fe/f-Butyl-4-{[(6-brom-3-methyl-2-phenylcN
butanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (146 mg, 428 μηηοΙ, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (1.5 ml) wurden HATU (244 mg, 642 μηηοΙ) und DIPEA (220 μΙ, 1.3 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-4-amino-3-(4-methylphenyl)butanoat (160 mg, 642 μηηοΙ), Beispiel 98A), gelöst in DMF (1 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60°C gerührt. Nach Abkühlen auf RT wurde das Gemisch mittels präparativer HPLC (Methode 21 ) gereinigt. Es wurden 138 mg (98% Reinheit, 55% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.54 min; MS (ESIpos): m/z = 573/575 [M+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.80 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.78-7.57 (br. s, 1 H), 7.56-7.45 (m, 5H), 7.21 (d, 2H), 7.13 (d, 2H), 3.89-3.40 (br. m, 2H, verdeckt), 3.39-3.26 (m, 1 H), 2.71 (dd, 1 H), 2.50 (1 H, verdeckt), 2.26 (s, 3H), 2.14 (br. s, 3H), 1.27 (s, 9H).
Beispiel 192A
(+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(3-methylphenyl)- butanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (1 18 mg, 345 μηηοΙ, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (1.0 ml) wurden HATU (197 mg, 518 μηηοΙ) und DIPEA (180 μΙ, 1.0 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-4-amino-3-(3-methylphenyl)butanoat (170 mg, 76% Reinheit, 518 μηηοΙ, Beispiel 99A), gelöst in DMF (1.0 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60°C gerührt. Nach Abkühlen auf RT wurde das Gemisch mittels praparativer HPLC (Methode 21 ) gereinigt. Es wurden 128 mg (98% Reinheit, 63% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.54 min; MS (ESIpos): m/z = 573/575 [M+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.81 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.80-7.58 (br. s, 1 H), 7.56-7.46 (m, 5H), 7.24-7.00 (m, 4H), 3.80-3.52 (br. m, 2H, verdeckt), 3.40-3.28 (m, 1 H), 2.72 (dd, 1 H), 2.54-2.45 (1 H, verdeckt), 2.27 (s, 3H), 2.13 (br. s, 3H), 1.26 (s, 9H).
Beispiel 193A
(+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-methoxyphenyl)- butanoat (Racemat)

Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (3.32 g, 9.71 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (35 ml) wurden HATU (5.54 g, 14.6 mmol) und DIPEA (5.1 ml, 29 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-4-amino-3-(2-methoxyphenyl)butanoat (4.20 g, 92% Reinheit, 14.6 mmol, Beispiel 100A), gelöst in DMF (20 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60°C gerührt. Nach Abkühlen auf RT wurde das Gemisch eingeengt, und der Rückstand wurde in Ethylacetat und Wasser (jeweils 200 ml) aufgenommen und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat extrahiert, und die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung (200 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und einrotiert. Der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (340 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat 97:3— > 8:2, Isolera One) gereinigt. Es wurden 4.50 g (98% Reinheit, 77% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.50 min; MS (ESIpos): m/z = 589/591 [M+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.77 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.71 (br. s, 1 H), 7.57-7.45 (m, 5H), 7.27-7.17 (m, 2H), 6.98 (d, 1 H), 6.91 (t, 1 H), 3.80 (s, 3H), 3.78-3.61 (m, 3H), 2.71-2.58 (m, 2H), 2.15 (s, 3H), 1.25 (s, 9H).
Beispiel 194A
(+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-[2-(trifluormethoxy)- phenyl]butanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (475 mg, 1.39 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (4.5 ml) wurden HATU (792 mg, 2.08 mmol) und DIPEA (730 μΙ, 4.2 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-4-amino-3-[2-(trifluormethoxy)phenyl]butanoat (700 mg, 95% Reinheit, 2.08 mmol, Beispiel 101A), gelöst in DMF (3 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60°C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethyl-
acetat und Wasser (jeweils 20 ml) versetzt und geschüttelt. Die wässrige Phase wurde zweimal mit Ethylacetat (20 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulen- Chromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat 85:15) gereinigt. Es wurden 367 mg (98% Reinheit, 40% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.60 min; MS (ESIpos): m/z = 643/645 [M+H]+
1H-NMR (600 MHz, DMSO-d6): δ [ppm] = 8.91 (t, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.81 -7.61 (br. m, 1 H), 7.61 -7.47 (m, 6H), 7.43-7.30 (m, 3H), 3.85-3.70 (m, 2H), 3.69-3.57 (m, 1 H), 2.76 (dd, 1 H), 2.60 (dd, 1 H), 2.16 (br. s, 3H), 1.26 (s, 9H).
Beispiel 195A
(+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(pyridin-2-yl)- butanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (425 mg, 1.24 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (4.5 ml) wurden HATU (708 mg, 1.86 mmol) und DIPEA (650 μΙ, 3.7 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-4-amino-3-(pyridin-2-yl)butanoat (550 mg, 80% Reinheit, 1.86 mmol, Beispiel 102A), gelöst in DMF (2 ml) hinzugegeben, und das Gemisch wur- de über Nacht bei 60°C gerührt. Nach Abkühlen auf RT wurde das Gemisch mittels präparativer HPLC (Methode 21 ) gereinigt. Es wurden 659 mg (98% Reinheit, 93% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.16 min; MS (ESIpos): m/z = 560/562 [M+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.88 (t, 1 H), 8.69 (d, 1 H), 8.09-7.98 (m, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.80-7.57 (br. m, 2H), 7.57-7.46 (m, 6H), 3.91-3.78 (m, 1 H), 3.75-3.58 (m, 2H), 2.89-2.72 (m, 2H), 2.14 (br. s, 3H), 1.28 (s, 9H).
Beispiel 196A
(+/-)-fe/f-Butyl-4-{[(6-brom-3-methyl-2-phenylcN
phenyl]butanoat (Racemat)

Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (857 mg, 2.51 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (10 ml) wurden HATU (1.43 g, 3.76 mmol) und DIPEA (1.3 ml, 7.5 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-4-amino-3-[2-(trifluormethyl)phenyl]butanoat (1.90 g, 60% Reinheit, 3.76 mmol, Beispiel 103A), gelöst in DMF (3 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Anschließend wurde das Gemisch mit Ethylacetat und Wasser (jeweils 20 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase zweimal mit Ethylacetat (jeweils 20 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulen- Chromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/ Ethylacetat 85:15, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert. Es wurden 1.10 g (98% Reinheit, 68% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.58 min; MS (ESIpos): m/z = 627/629 [M+H]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.249 (16.00), 1.397 (4.44), 2.161 (1.10), 2.687 (0.40), 2.706 (0.41 ), 2.735 (0.40), 2.751 (0.41 ), 3.314 (1.68), 7.474 (0.59), 7.492 (0.67), 7.506 (1.12), 7.525 (1.58), 7.539 (2.10), 7.558 (0.44), 7.685 (0.56), 7.718 (0.64), 7.738 (0.55), 7.760 (0.65), 7.779 (0.41 ), 7.838 (0.41 ), 7.861 (0.63), 7.865 (0.57), 7.939 (1.09), 7.962 (0.71 ), 8.902 (0.54).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.90 (t, 1 H), 7.95 (d, 1 H), 7.85 (dd, 1 H), 7.79-7.57 (m, 4H), 7.57-7.44 (m, 6H), 3.90-3.60 (m, 3H), 2.75 (dd, 1 H), 2.69 (dd, 1 H), 2.16 (br. s, 3H), 1.25 (s, 9H).
Beispiel 197A
(+/-)-fe/f-Butyl-4-{[(6-brom-3-methyl-2-phenylcN
2-yl)butanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (1.64 g, 4.79 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (20 ml) wurden HATU (2.73 g, 7.19 mmol) und DIPEA (2.5 ml, 14 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ierf-Butyl-4-amino-3-(3-methoxypyridin-2-yl)butanoat (2.02 g, 95% Reinheit, 7.19 mmol, Beispiel 104A), gelöst in DMF (8 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60°C gerührt. Nach Abkühlen auf RT wurde das Gemisch mittels präpara- tiver HPLC (Methode 21 ) gereinigt. Es wurden 1.73 g (98% Reinheit, 60% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.24 min; MS (ESIpos): m/z = 590/592 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.255 (16.00), 2.192 (4.16), 5.754 (0.86), 7.506 (0.89), 7.51 1 (0.69), 7.524 (1.44), 7.536 (1.73), 7.538 (1.71 ), 7.543 (1.00), 7.546 (0.66), 7.843 (0.42), 7.865 (0.65), 7.871 (0.58), 7.937 (1.13), 7.959 (0.71 ), 8.128 (0.53), 8.131 (0.53), 8.140 (0.54).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.77 (t, 1 H), 8.14 (dd, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.77 (s, 1 H), 7.58-7.47 (m, 5H), 7.43 (d, 1 H), 7.27 (dd, 1 H), 4.02-3.90 (m, 1 H), 3.84 (s, 3H), 3.79- 3.62 (m, 2H), 2.87 (dd, 1 H), 2.63 (dd, 1 H), 2.19 (s, 3H), 1.25 (s, 9H). Beispiel 198A
(+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-chlorphenyl)-2,2- dimethylbutanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (306 mg, 895 μηηοΙ, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (1.5 ml) wurden HATU (443 mg, 1.16 mmol) und DIPEA (470 μΙ, 2.7 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-4-amino-3-(2-chlorphenyl)-2,2-dimethylbutanoat (320 mg, 1.07 mmol, Beispiel 106A), gelöst in DMF (1 ml) hinzugegeben, und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch mittels präparativer HPLC (Methode: Chromatorex C18, 10 μηι, 30 x 125 mm, Acetonitril / Wasser-Gradient: Acetonitril 10%— > 90%) gereinigt. Es wurden 328 mg (99% Reinheit, 59% d. Th.) der Titelverbindung erhalten. LC-MS (Methode 2): Rt = 1.53 min; MS (ESIpos): m/z = 621/523 (M+H)+
1 H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.69 (t, 1 H), 7.90 (d, 1 H), 7.82 (dd, 1 H), 7.58-7.22 (m, 10H), 4.17-4.09 (m, 1 H), 4.05-3.87 (m, 1 H), 3.77-3.63 (m, 1 H), 1.97 (br. s, 3H), 1.48 (s, 9H), 1.10 (s, 3H), 1.06 (s, 3H).
Beispiel 199A
(+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-chlorphenyl)-3- methylbutanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (1.13 g, 3.29 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (12 ml) wurden HATU (1.88 g,
4.93 mmol) und DIPEA (1.7 ml, 9.9 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ierf-Butyl-4-amino-3-(2-chlorphenyl)-3-methylbutanoat (2.00 g, 70% Reinheit, 4.93 mmol, Beispiel 107A), gelöst in DMF (6 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60°C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethylacetat und Wasser (jeweils 50 ml) versetzt und geschüttelt. Die wässrige Phase wurde zweimal mit Ethylacetat (50 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässri- ger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat 85:15) vorgereinigt. Es wurden 1.94 g (87% Reinheit, 84% d. Th.) der vorgereinigten Titelverbindung erhalten. Von diesen
1.94 g wurden 250 mg mittels präparativer HPLC (Methode 20) nachgereinigt. Es wurden daraus 144 mg (98% Reinheit) der nachgereinigten Titelverbindung (siehe Analytik) erhalten.
LC-MS (Methode 1 ): Rt = 2.63 min; MS (ESIpos): m/z = 607/609 (M+H)+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.1 19 (16.00), 1.652 (2.39), 2.572 (0.66), 2.608 (0.71 ), 3.415 (0.53), 3.450 (0.49), 7.287 (0.59), 7.305 (0.42), 7.433 (0.75), 7.452 (0.44), 7.457 (0.42), 7.499 (0.89), 7.506 (0.75), 7.518 (1.75), 7.527 (1.66), 7.853 (0.53), 7.857 (0.52), 7.927 (0.99), 7.950 (0.63), 8.664 (0.43).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.66 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.65 (br. s, 1 H), 7.57-7.47 (m, 5H), 7.46-7.37 (m, 2H), 7.33-7.23 (m, 2H), 4.37 (dd, 1 H), 3.75 (dd, 1 H), 3.43 (d, 1 H), 2.59 (d, 1 H), 2.12 (br. s, 3H), 1.65 (s, 3H), 1.12 (s, 9H).
Beispiel 200A
(+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-fluorphenyl)-3- methylbutanoat (Racemat)

Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (612 mg, 1.79 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (7 ml) wurden HATU (1.02 g, 2.68 mmol) und DIPEA (940 μΙ, 5.4 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-4-amino-3-(2-fluorphenyl)-3-methylbutanoat (810 mg, „2.68 mmol", nicht reinheitskorrigiert, Beispiel 108A), gelöst in DMF (3 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethylacetat und Wasser (jeweils 50 ml) versetzt und geschüttelt. Die wässrige Phase wurde zweimal mit Ethylacetat (50 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und einge- engt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat 85:15) vorgereinigt. Das vorgereinigte Produkt wurde mittels präparativer HPLC (Methode 20) nachgereinigt. Es wurden 255 mg (95% Reinheit,„23% d. Th.") der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.55 min; MS (ESIpos): m/z = 591/593 (M+H)+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.126 (16.00), 1.197 (0.85), 1.569 (2.52), 2.976 (0.56), 3.01 1 (0.50), 7.139 (0.43), 7.157 (0.47), 7.172 (0.41 ), 7.315 (0.52), 7.335 (0.60), 7.502 (0.97), 7.509 (0.83), 7.520 (1.94), 7.529 (1.82), 7.857 (0.57), 7.862 (0.59), 7.931 (1.10), 7.954 (0.69), 8.726 (0.42).
Trennung der Enantiomere:
Die Titelverbindung (220 mg) wurde in einem Gemisch aus Isopropanol (4 ml) und Dichlormethan (2 ml) gelöst und mittels präparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 201A und 202A) [Säule: Daicel Chiralpak IE, 5 μηι, 250 mm x 20 mm; Fluss: 15 ml/min; Detektion: 220 nm; Temperatur: 40 °C; Injektion: 0.2 ml; Eluent: 60% Heptan / 40% Isopropanol; isokratisch; Laufzeit 20 min]. Beispiel 201 A
(+)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-fluorphenyl)-3- methylbutanoat (Enantiomer 1)
Bei der in Beispiel 200A beschriebenen Enantiomerentrennung wurden 96 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als früher eluierendes Enantiomer erhalten. [a]D 20 = +20.7°, 589 nm, c = 0.25 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.35 min; MS (ESIpos): m/z = 591/593 (M+H)+
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: 1.126 (16.00), 1.197 (0.42), 1.569 (2.02), 2.979 (0.44), 3.007 (0.41 ), 7.335 (0.41 ), 7.501 (0.66), 7.505 (0.68), 7.517 (1.25), 7.529 (1.41 ), 7.852 (0.41 ), 7.856 (0.41 ), 7.930 (0.88), 7.948 (0.60).
1H-NMR (500 MHz, DMSO-d6): δ [ppm] = 8.72 (t, 1 H), 7.94 (d, 1 H), 7.84 (dd, 1 H), 7.68 (br. s, 1 H), 7.56-7.47 (m, 5H), 7.37-7.28 (m, 2H), 7.19-7.10 (m, 2H), 4.08 (dd, 1 H), 3.62 (dd, 1 H), 2.99 (d, 1 H), -2.5 (1 H, verdeckt), 2.08 (br. s, 3H), 1.57 (s, 3H), 1.13 (s, 9H).
Beispiel 202A
(-)-ie f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]ami
methylbutanoat (Enantiomer 2)
Bei der in Beispiel 200A beschriebenen Enantiomerentrennung wurden 97 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = -17.2°, 589 nm, c = 0.25 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.35 min; MS (ESIpos): m/z = 591/593 (M+H)+
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: 1.126 (16.00), 1.569 (2.16), 2.979 (0.47), 3.007 (0.43), 7.335 (0.45), 7.501 (0.71 ), 7.504 (0.72), 7.517 (1.33), 7.528 (1.50), 7.851 (0.44), 7.855 (0.42), 7.930 (0.90), 7.948 (0.61 ).
1H-NMR (500 MHz, DMSO-d6): δ [ppm] = 8.72 (t, 1 H), 7.94 (d, 1 H), 7.84 (dd, 1 H), 7.65 (br. s, 1 H), 7.56-7.45 (m, 5H), 7.37-7.26 (m, 2H), 7.20-7.07 (m, 2H), 4.08 (dd, 1 H), 3.62 (dd, 1 H), 2.99 (d, 1 H), -2.5 (1 H, verdeckt), 2.10 (br. s, 3H), 1.57 (s, 3H), 1.17-1.07 (m, 9H).
Beispiel 203A
(+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-methyl-3-(pyridin-2- yl)butanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (2.40 g, 7.02 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (30 ml) wurden HATU (4.01 g, 10.5 mmol) und DIPEA (3.7 ml, 21 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde das in Beispiel 109A erhaltene Gemisch (4.00 g) aus (+/-)-tert- Butyl-4-amino-3-methyl-3-(pyridin-2-yl)butanoat und (+/-)-ie/f-Butyl-4-amino-3-(3-chlorpyridin-2-
yl)-3-methylbutanoat, gelöst in DMF (10 ml) hinzugegeben, und das Reaktionsgemisch wurde über Nacht bei 60°C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethylacetat und Wasser (jeweils 50 ml) versetzt, geschüttelt, und die Phasen wurden getrennt. Die wässrige Phase wurde zweimal mit Ethylacetat (50 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriet und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohe- xan/Ethylacetat 85:15) vorgereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert. Anschließend wurde mittels präparativer HPLC (Methode 20) nachgerei- nigt. Es wurden 1.07 g (98% Reinheit) der Titelverbindung, sowie 1.56 g (98% Reinheit) ie/f- Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(3-chlorpyridin-2-yl)-3- methylbutanoat erhalten.
LC-MS (Methode 1 ): Rt = 2.37 min; MS (ESIpos): m/z = 574/576 [M+H]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.180 (1.07), 1.205 (16.00), 1.552 (2.74), 2.141 (0.81 ), 2.159 (0.47), 2.610 (0.47), 2.647 (0.55), 3.049 (0.44), 3.162 (0.58), 7.499 (0.90), 7.506 (0.72), 7.517 (1.45), 7.529 (1.88), 7.836 (0.42), 7.858 (0.63), 7.864 (0.59), 7.933 (1.25), 7.955 (0.91 ).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.73 (br. t, 1 H), 8.64 (br. d, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.69-7.61 (m, 2H), 7.57-7.47 (m, 6H), 7.46-7.38 (m, 1 H), 3.96 (dd, 1 H), 3.76 (dd, 1 H), 3.08 (d, 1 H), 2.64 (d, 1 H), 2.16 (s, 3H), 1.56 (s, 3H), 1.21 (s, 9H). Beispiel 204A
(+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-methoxyphenyl)- 3-methylbutanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (3.76 g, 1 1.0 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (45 ml) wurden HATU (6.26 g, 16.5 mmol) und DIPEA (5.7 ml, 33 mmol) gegeben und das Gemisch wurde 30 min bei RT
gerührt. Anschließend wurde (+/-)-ie/f-Butyl-4-amino-3-(2-methoxyphenyl)-3-methylbutanoat (5.00 g, 92% Reinheit, 16.5 mmol, Beispiel 1 10A), gelöst in DMF (20 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60°C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethylacetat und Wasser (jeweils 50 ml) versetzt und geschüttelt. Die wässrige Phase wurde zweimal mit Ethylacetat (50 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat 93:7 — > 6:4, Isolera One) gereinigt. Es wurden 6.00 g (98% Reinheit, 89% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 7): Rt = 1.70 min; MS (ESIpos): m/z = 603/605 (M+H)+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.52), 0.008 (0.45), 1.095 (16.00), 1.398 (6.40), 1.553 (2.19), 2.433 (0.64), 2.466 (0.72), 3.181 (0.56), 3.214 (0.53), 3.309 (5.17), 6.889 (0.46), 6.986 (0.41 ), 7.006 (0.48), 7.188 (0.55), 7.208 (0.67), 7.495 (0.76), 7.503 (0.70), 7.514 (1.98), 7.518 (1.79), 7.527 (1.04), 7.848 (0.57), 7.853 (0.55), 7.921 (1.07), 7.944 (0.68).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.54 (t, 1 H), 7.93 (d, 1 H), 7.84 (dd, 1 H), 7.70 (br. s, 1 H), 7.56-7.44 (m, 5H), 7.27-7.14 (m, 2H), 7.00 (d, 1 H), 6.89 (t, 1 H), 4.18 (dd, 1 H), 3.68 (dd, 1 H), 3.20 (d, 1 H), 2.45 (d, 1 H), 2.10 (br. s, 3H), 1.55 (s, 3H), 1.09 (s, 9H).
Trennung der Enantiomere:
Die Titelverbindung (2.0 g) wurde in einem Gemisch (100 ml) aus Methanol und Acetonitril gelöst und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 205A und 206A) [Säule: Daicel Chiralpak AD-H, 5 μηι, 250 mm x 30 mm; Fluss: 150 ml/min; De- tektion: 210 nm; Temperatur: 38 °C; Injektion: 0.4 ml; Eluent: 75% Kohlendioxid / 25% Ethanol; isokratisch; Laufzeit 6.5 min]. Beispiel 205A
(+)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-methoxyphenyl)-3- methylbutanoat (Enantiomer 1)
Bei der in Beispiel 204A beschriebenen Enantiomerentrennung wurden 723 mg (98% Reinheit, ee-Wert 94%) der Titelverbindung als früher eluierendes Enantiomer erhalten. [a]D 20 = +16.2°, 589 nm, c = 0.50 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.60 min; MS (ESIpos): m/z = 603/605 [M+H]+
1H-NMR (600 MHz, DMSO-d6) δ [ppm]: 1.091 (16.00), 1.555 (2.18), 2.434 (0.67), 2.456 (0.71 ), 3.193 (0.51 ), 3.215 (0.49), 3.858 (3.12), 7.191 (0.53), 7.202 (0.46), 7.204 (0.48), 7.498 (0.54), 7.502 (0.74), 7.515 (1.28), 7.525 (1.65), 7.848 (0.47), 7.851 (0.46), 7.928 (0.94), 7.942 (0.69).
1H-NMR (600 MHz, DMSO-d6): δ [ppm] = 8.56 (t, 1 H), 7.93 (d, 1 H), 7.84 (dd, 1 H), 7.68 (br. s, 1 H), 7.57-7.43 (m, 5H), 7.28-7.16 (m, 2H), 7.00 (d, 1 H), 6.89 (t, 1 H), 4.19 (dd, 1 H), 3.67 (dd, 1 H), 3.20 (d, 1 H), 2.45 (d, 1 H), 2.09 (br. s, 3H), 1.56 (s, 3H), 1.09 (s, 9H).
Beispiel 206A
(-)-ie f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]ami
methylbutanoat (Enantiomer 2)
Bei der in Beispiel 204A beschriebenen Enantiomerentrennung wurden 718 mg (98% Reinheit, ee-Wert 100%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = -19.2°, 589 nm, c = 0.25 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.60 min; MS (ESIpos): m/z = 603/605 [M+H]+
1H-NMR (600 MHz, DMSO-d6) δ [ppm]: 1.091 (16.00), 1.556 (2.09), 2.435 (0.66), 2.456 (0.70), 3.193 (0.49), 3.215 (0.48), 3.350 (1.37), 7.189 (0.45), 7.191 (0.51 ), 7.202 (0.44), 7.204 (0.47), 7.497 (0.53), 7.501 (0.72), 7.514 (1.23), 7.525 (1.56), 7.847 (0.46), 7.851 (0.44), 7.927 (0.96), 7.942 (0.69).
1H-NMR (600 MHz, DMSO-d6): δ [ppm] = 8.56 (t, 1 H), 7.93 (d, 1 H), 7.84 (dd, 1 H), 7.67 (br. s, 1 H), 7.56-7.43 (m, 5H), 7.28-7.15 (m, 2H), 7.00 (d, 1 H), 6.89 (t, 1 H), 4.19 (dd, 1 H), 3.67 (dd, 1 H), 3.20 (d, 1 H), 2.45 (d, 1 H), 2.13 (br. s, 1 H), 1.56 (s, 3H), 1.09 (s, 9H).
Beispiel 207A
(+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(5-fluor-2-methoxy- phenyl)butanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (86 mg, 250 μηηοΙ, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (2 ml) wurden HATU (143 mg, 375 μηηοΙ) und DIPEA (130 μΙ, 750 μηηοΙ) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-4-amino-3-(5-fluor-2-methoxyphenyl)butanoat-Hydrochlorid
(120 mg, 375 μηηοΙ, Beispiel 1 11A), gelöst in DMF (1 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60°C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethylacetat und Wasser (jeweils 50 ml) versetzt und geschüttelt. Die wässrige Phase wurde zweimal mit Ethylacetat (50 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/ Ethylacetat 93:7— > 6:4, Isolera One) gereinigt. Es wurden 100 mg (98% Reinheit, 64% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.49 min; MS (ESIpos): m/z = 607/609 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.235 (0.42), 1.266 (16.00), 2.145 (1.72), 2.621 (0.48), 2.641 (0.86), 2.654 (0.51 ), 3.741 (0.60), 3.753 (0.58), 3.779 (4.38), 6.990 (0.70), 7.003 (0.83), 7.084 (0.46), 7.091 (0.40), 7.108 (0.45), 7.503 (1.00), 7.510 (0.84), 7.522 (2.12), 7.529 (2.02), 7.837 (0.43), 7.859 (0.65), 7.864 (0.61 ), 7.933 (1.19), 7.956 (0.75), 8.783 (0.51 ).
Trennung der Enantiomere:
Die Titelverbindung (440 mg, aus einem analog durchgeführten Wiederholexperiment) wurde in einem Gemisch aus Acetonitril (2 ml) und Isopropanol (2 ml) gelöst und mittels präparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 208A und 209A) [Säule: Daicel Chiralpak ID, 5 μηη, 250 mm x 20 mm; Fluss: 20 ml/min; Detektion: 220 nm; Temperatur: 23 °C; Injektion: 0.1 ml; Eluent: 80% Heptan / 20% Ethanol; isokratisch]. Beispiel 208A
(-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(5-fluor-2-methoxy- phenyl)butanoat (Enantiomer 1)
Bei der in Beispiel 207A beschriebenen Enantiomerentrennung wurden 181 mg (98% Reinheit, ee-Wert 98%) der Titelverbindung als früher eluierendes Enantiomer erhalten. [a]D 20 = -6.4°, 589 nm, c = 0.37 g/100 ml, Chloroform
LC-MS (Methode 1 ): Rt = 2.49 min; MS (ESIpos): m/z = 607/609 [M+H]+
1H-NMR (600 MHz, DMSO-d6) δ [ppm]: 1.265 (16.00), 2.146 (0.51 ), 2.688 (0.46), 6.992 (0.51 ), 7.000 (0.48), 7.506 (0.56), 7.51 1 (0.60), 7.521 (0.43), 7.524 (1.24), 7.534 (1.92), 7.537 (1.48), 7.862 (0.48), 7.865 (0.45), 7.940 (0.98), 7.955 (0.72), 8.804 (0.41 ).
1H-NMR (600 MHz, DMSO-d6): δ [ppm] = 8.80 (t, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.66 (br. s, 1 H), 7.57-7.46 (m, 5H), 7.1 1 (dd, 1 H), 7.03 (td, 1 H), 6.99 (dd, 1 H), 3.79-3.70 (m, 2H), 3.78 (s, 3H), 3.68-3.62 (m, 1 H), 2.69-2.60 (m, 2H), 2.15 (br. s, 3H), 1.27 (s, 9H).
Beispiel 209A
(+)-fe/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchm^
methoxyphenyl)butanoat (Enantiomer 2)
Bei der in Beispiel 207A beschriebenen Enantiomerentrennung wurden 175 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = +2.9°, 589 nm, c = 0.38 g/100 ml, Chloroform
LC-MS (Methode 1 ): Rt = 2.49 min; MS (ESIpos): m/z = 607/609 [M+H]+
1H-NMR (600 MHz, DMSO-d6) δ [ppm]: 1.266 (16.00), 2.146 (0.52), 6.992 (0.51 ), 7.000 (0.48), 7.506 (0.55), 7.51 1 (0.60), 7.521 (0.40), 7.524 (1.24), 7.534 (1.92), 7.537 (1.52), 7.862 (0.48), 7.865 (0.46), 7.940 (0.97), 7.955 (0.71 ), 8.805 (0.42).
1H-NMR (600 MHz, DMSO-d6): δ [ppm] = 8.80 (t, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.66 (br. s, 1 H), 7.56-7.47 (m, 5H), 7.1 1 (dd, 1 H), 7.03 (td, 1 H), 6.99 (dd, 1 H), 3.78 (s, 3H), 3.77-3.70 (m, 2H), 3.68-3.63 (m, 1 H), 2.69-2.59 (m, 2H), 2.15 (br. s, 3H), 1.27 (s, 9H).
Beispiel 210A
(+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-fluor-6-methoxy- phenyl)butanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (583 mg, 1.70 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (6 ml) wurden HATU (778 mg, 2.04 mmol) und DIPEA (710 μΙ, 4.1 mmol) gegeben und das Gemisch wurde 15 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-4-amino-3-(2-fluor-6-methoxyphenyl)butanoat (587 mg, 82% Reinheit, 1.70 mmol, Beispiel 1 12A), gelöst in DMF (5 ml) hinzugegeben, und das Gemisch wurde 5 h bei 60 °C und danach über Nacht bei RT gerührt. Anschließend wurde das Gemisch mit Ethylacetat und Wasser (jeweils 100 ml) versetzt und geschüttelt. Die wässrige Phase wurde einmal mit Ethylacetat (80 ml) extrahiert. Die vereinigten organischen Phasen wur-
den mit gesättigter wässriger Natriumchlorid-Lösung (100 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohe- xan/Ethylacetat 93:7 -> 7:3, Isolera One) gereinigt. Es wurden 441 mg (100% Reinheit, 43% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.49 min; MS (ESIpos): m/z = 607/609 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.175 (0.67), 1.192 (0.45), 1.225 (16.00), 1.248 (0.75), 1.988 (1.21 ), 2.169 (0.92), 2.683 (0.52), 2.697 (0.66), 3.815 (4.33), 6.761 (0.45), 6.835 (0.52), 6.855 (0.58), 7.503 (0.81 ), 7.510 (0.71 ), 7.522 (1.95), 7.527 (1.89), 7.535 (1.21 ), 7.840 (0.42), 7.863 (0.64), 7.868 (0.60), 7.933 (1.20), 7.955 (0.74), 8.860 (0.46).
1 H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.86 (t, 1 H), 7.94 (d, 1 H), 7.86 (dd, 1 H), 7.72 (br. s, 1 H), 7.56-7.47 (m, 5H), 7.29-7.16 (m, 1 H), 6.85 (d, 1 H), 6.76 (t, 1 H), 3.99-3.88 (m, 1 H), 3.82 (s, 3H), 3.84-3.64 (m, 2H), 2.76-2.62 (m, 2H), 2.17 (br. s, 3H), 1.23 (s, 9H).
Trennung der Enantiomere:
Die Titelverbindung (437 mg) wurde in Methanol (35 ml) gelöst und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 21 1A und 212A) [Säule: Daicel Chi- ralpak AD SFC, 250 mm x 20 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 0.3 ml; Eluent: 80% Kohlendioxid / 20% Ethanol; isokratisch, Laufzeit 8.0 min].
Beispiel 211A
(+)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-fluor-6-methoxy- phenyl)butanoat (Enantiomer 1)
Bei der in Beispiel 210A beschriebenen Enantiomerentrennung wurden 203 mg (100% Reinheit, ee-Wert 100%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = +12.9°, 589 nm, c = 0.39 g/100 ml, Chloroform
LC-MS (Methode 2): Rt = 1.31 min; MS (ESIpos): m/z = 607/609 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.225 (16.00), 2.170 (1.08), 2.684 (0.57), 2.698 (0.77), 2.719 (0.41 ), 3.815 (4.50), 6.762 (0.50), 6.834 (0.57), 6.855 (0.63), 7.503 (0.85), 7.510 (0.73), 7.522 (2.02), 7.528 (1.96), 7.862 (0.62), 7.867 (0.60), 7.933 (1.1 1 ), 7.955 (0.69), 8.861 (0.53).
1 H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.86 (t, 1 H), 7.94 (d, 1 H), 7.86 (dd, 1 H), 7.72 (br. s, 1 H), 7.57-7.46 (m, 5H), 7.24 (dd, 1 H), 6.85 (d, 1 H), 6.76 (t, 1 H), 4.01 -3.87 (m, 1 H), 3.82 (s, 3H), 3.83-3.65 (m, 2H), 2.77-2.61 (m, 2H), 2.17 (br. s, 3H), 1.23 (s, 9H).
Beispiel 212A
(-)-fe/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4^
phenyl)butanoat (Enantiomer 2)
Bei der in Beispiel 21 OA beschriebenen Enantiomerentrennung wurden 185 mg (99% Reinheit, ee-Wert 96%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = -14.2°, 589 nm, c = 0.44 g/100 ml, Chloroform
LC-MS (Methode 2): Rt = 1.31 min; MS (ESIpos): m/z = 607/609 [M+H]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.225 (16.00), 1.249 (0.71 ), 2.170 (1.46), 2.684 (0.70), 2.698 (0.99), 2.720 (0.52), 3.314 (6.09), 3.719 (0.48), 3.796 (0.41 ), 6.762 (0.63), 6.785 (0.40), 6.834 (0.65), 6.855 (0.72), 7.226 (0.42), 7.503 (1.06), 7.508 (0.96), 7.527 (2.63), 7.843 (0.46), 7.866 (0.74), 7.933 (1.12), 7.955 (0.70), 8.861 (0.66).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.86 (t, 1 H), 7.94 (d, 1 H), 7.86 (d, 1 H), 7.72 (br. s, 1 H), 7.56-7.47 (m, 5H), 7.24 (dd, 1 H), 6.85 (d, 1 H), 6.76 (t, 1 H), 4.01 -3.88 (m, 1 H), 3.82 (s, 3H), 3.85- 3.65 (m, 2H), 2.78-2.62 (m, 2H), 2.17 (br. s, 3H), 1.23 (s, 9H).
Beispiel 213A
(+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-ethoxyphenyl)- butanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (274 mg, 802 μηηοΙ, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (3 ml) wurden HATU (457 mg, 1.20 mmol) und DIPEA (420 μΙ, 2.4 mmol) gegeben und das Gemisch wurde 15 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-4-amino-3-(2-ethoxyphenyl)butanoat-Hydrochlorid (400 mg, 95% Reinheit, 1 .20 mmol, Beispiel 1 13A), gelöst in DMF (2 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Anschließend wurde das Gemisch mit Ethylacetat und Wasser (jeweils 20 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige
Phase zweimal mit Ethylacetat (20 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohe- xan/Ethylacetat 85:15, Isolera One) gereinigt. Es wurden 265 mg (98% Reinheit, 54% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.62 min; MS (ESIpos): m/z = 603/605 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.245 (16.00), 1.356 (1.23), 1.373 (2.56), 1.391 (1.23), 2.151 (1.35), 2.661 (0.70), 2.675 (0.57), 2.682 (0.48), 3.660 (0.41 ), 4.040 (0.88), 4.058 (0.84), 6.884 (0.57), 6.954 (0.52), 6.974 (0.58), 7.193 (1.00), 7.212 (0.79), 7.502 (0.86), 7.509 (0.79), 7.521 (1.77), 7.527 (1.71 ), 7.530 (1.45), 7.534 (1.09), 7.538 (0.84), 7.836 (0.41 ), 7.858 (0.62), 7.864 (0.56), 7.932 (1.09), 7.955 (0.69).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.76 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.72 (br. s, 1 H), 7.56-7.45 (m, 5H), 7.23-7.14 (m, 2H), 6.96 (d, 1 H), 6.88 (td, 1 H), 4.05 (q, 2H), 3.87-3.75 (m, 1 H), 3.73-3.58 (m, 2H), 2.71 -2.63 (m, 2H), 2.15 (br. s, 3H), 1.37 (t, 3H), 1.25 (s, 9H).
Trennung der Enantiomere:
Die Titelverbindung (240 mg) wurde in Methanol (20 ml) gelöst und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 214A und 215A) [Säule: Daicel Chi- ralcel OJ-H, 250 mm x 20 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injekti- on: 0.5 ml; Eluent: 83% Kohlendioxid / 17% Ethanol; isokratisch, Laufzeit 8.0 min].
Beispiel 214A
(-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-ethoxyphenyl)- butanoat (Enantiomer 1)
Bei der in Beispiel 213A beschriebenen Enantiomerentrennung wurden 79 mg (98% Reinheit, ee-Wert 100%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -17.1 °, 589 nm, c = 0.35 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.39 min; MS (ESIpos): m/z = 603/605 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.245 (16.00), 1.356 (1.21 ), 1.373 (2.58), 1.391 (1.25), 2.151 (1.59), 2.661 (0.78), 2.676 (0.65), 3.660 (0.47), 4.040 (0.94), 4.057 (0.93), 6.885 (0.65), 6.954 (0.58), 6.974 (0.67), 7.193 (1.06), 7.212 (0.83), 7.502 (0.89), 7.508 (0.75), 7.521 (1.93), 7.527 (1.92), 7.836 (0.43), 7.858 (0.64), 7.863 (0.61 ), 7.932 (1.18), 7.955 (0.75), 8.764 (0.43).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.77 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.72 (br. s, 1 H), 7.57-7.45 (m, 5H), 7.23-7.14 (m, 2H), 6.96 (d, 1 H), 6.89 (t, 1 H), 4.05 (q, 2H), 3.88-3.77 (m, 1 H), 3.74-3.58 (m, 2H), 2.73-2.60 (m, 2H), 2.15 (br. s, 3H), 1.37 (t, 3H), 1.25 (s, 9H).
Beispiel 215A
(+)-ie f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]ami
butanoat (Enantiomer 2)
Bei der in Beispiel 213A beschriebenen Enantiomerentrennung wurden 72 mg (98% Reinheit, ee-Wert 98%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = +10.9°, 589 nm, c = 0.34 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.39 min; MS (ESIpos): m/z = 603/605 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.245 (16.00), 1.356 (1.23), 1.374 (2.59), 1.391 (1.26), 2.151 (1.65), 2.661 (0.83), 2.676 (0.69), 3.661 (0.50), 4.040 (0.98), 4.058 (0.96), 6.885 (0.67), 6.954 (0.60), 6.974 (0.70), 7.193 (1.1 1 ), 7.212 (0.86), 7.502 (0.92), 7.509 (0.79), 7.521 (2.00), 7.528 (1.96), 7.836 (0.42), 7.858 (0.64), 7.863 (0.60), 7.932 (1.1 1 ), 7.955 (0.71 ), 8.764 (0.46). 1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.76 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.72 (br. s, 1 H), 7.58-7.46 (m, 5H), 7.27-7.12 (m, 2H), 6.96 (d, 1 H), 6.89 (t, 1 H), 4.05 (q, 2H), 3.88-3.75 (m, 1 H), 3.71-3.59 (m, 2H), 2.72-2.62 (m, 2H), 2.15 (br. s, 3H), 1.37 (t, 3H), 1.25 (s, 9H).
Beispiel 216A
(+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(5-fluor-2-methoxy- phenyl)-3-methylbutanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (812 mg, 2.37 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (8 ml) wurden HATU (1.35 g, 3.56 mmol) und DIPEA (1.2 ml, 7.1 mmol) gegeben und das Gemisch wurde 15 min bei RT
gerührt. Anschließend wurde (+/-)-ie/f-Butyl-4-amino-3-(5-fluor-2-methoxyphenyl)-3-methyl- butanoat-Hydrochlorid (1.25 g, 95% Reinheit, 3.56 mmol, Beispiel 1 14A), gelöst in DMF (5 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Anschließend wurde das Gemisch mit Ethylacetat und Wasser (jeweils 20 ml) versetzt und geschüttelt. Nach Phasentren- nung wurde die wässrige Phase zweimal mit Ethylacetat (20 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP- Sil, Cyclohexan/ Ethylacetat 85:15, Isolera One) gereinigt. Es wurden 930 mg (98% Reinheit, 62% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.57 min; MS (ESIpos): m/z = 621/623 [M+H]+
1H-NMR (600 MHz, DMSO-d6) δ [ppm]: 1.131 (16.00), 1.526 (2.14), 2.441 (0.62), 2.463 (0.66), 3.336 (6.15), 7.503 (0.52), 7.506 (0.69), 7.520 (1.18), 7.530 (1.38), 7.933 (0.73), 7.948 (0.54).
1H-NMR (600 MHz, DMSO-d6): δ [ppm] = 8.59 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.69 (br. s, 1 H), 7.56-7.47 (m, 5H), 7.09-6.99 (m, 2H), 6.97 (dd, 1 H), 4.24 (br. s, 1 H), 3.85 (s, 3H), 3.61 (br. d, 1 H), 3.21 (d, 1 H), 2.45 (d, 1 H), 2.1 1 (br. s, 3H), 1.53 (s, 3H), 1.13 (s, 9H).
Trennung der Enantiomere:
Die Titelverbindung (775 mg) wurde in Isopropanol (30 ml) aufgenommen, filtriert und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 217A und 218A) [Säule: Daicel Chiralpak AD, 360 mm x 50 mm; Fluss: 400 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 2.0 ml; Eluent: 75% Kohlendioxid / 25% Ethanol; isokratisch, Laufzeit 9.0 min]. Die vereinigten Zielfraktionen wurden eingeengt.
Beispiel 217A
(+)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(5-fluor-2-methoxy- phenyl)-3-methylbutanoat (Enantiomer 1)
Bei der in Beispiel 216A beschriebenen Enantiomerentrennung wurden 377 mg („98% Reinheit", Isopropanol-haltig, ee-Wert 96%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = +1 1.9°, 589 nm, c = 0.35 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.56 min; MS (ESIpos): m/z = 621/623 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.030 (5.33), 1.045 (5.34), 1.135 (16.00), 1.526 (2.20), 2.440 (0.63), 2.474 (0.74), 3.187 (0.48), 3.221 (0.45), 3.31 1 (4.42), 4.323 (0.65), 4.333 (0.62), 6.983 (0.48), 7.500 (0.81 ), 7.508 (0.72), 7.519 (1.81 ), 7.525 (1.58), 7.852 (0.52), 7.857 (0.51 ), 7.928 (1.03), 7.950 (0.65).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.57 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.68 (br. s, 1 H), 7.56-7.46 (m, 5H), 7.10-6.99 (m, 2H), 6.97 (dd, 1 H), 4.23 (dd, 1 H), 3.85 (s, 3H), 3.62 (dd, 1 H), 3.20 (d, 1 H), 2.46 (d, 1 H), 2.10 (br. s, 3H), 1.53 (s, 3H), 1.13 (s, 9H).
Beispiel 218A
(-)-ie f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(5-fl
phenyl)-3-methylbutanoat (Enantiomer 2)
Bei der in Beispiel 216A beschriebenen Enantiomerentrennung wurden 295 mg („98% Reinheit", Isopropanol-haltig, ee-Wert 99%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = -1 1.5°, 589 nm, c = 0.36 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.56 min; MS (ESIpos): m/z = 621/623 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.030 (4.50), 1.045 (4.53), 1.134 (16.00), 1.526 (2.35), 2.440 (0.64), 2.474 (0.74), 3.187 (0.51 ), 3.221 (0.47), 3.853 (4.12), 4.323 (0.65), 4.333 (0.62), 6.955 (0.42), 6.982 (0.50), 7.500 (0.85), 7.508 (0.76), 7.519 (1.92), 7.525 (1.68), 7.852 (0.53), 7.857 (0.52), 7.927 (1.00), 7.950 (0.64).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.57 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.68 (br. s, 1 H), 7.56-7.46 (m, 5H), 7.10-6.99 (m, 2H), 6.96 (dd, 1 H), 4.23 (dd, 1 H), 3.85 (s, 3H), 3.62 (dd, 1 H), 3.20 (d, 1 H), 2.46 (d, 1 H), 2.10 (br. s, 3H), 1.53 (s, 3H), 1.13 (s, 9H).
Beispiel 219A
(+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(3,5-difluor-2- methoxyphenyl)-3-methylbutanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (819 mg, 2.39 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (8 ml) wurden HATU (1.37 g, 3.59 mmol) und DIPEA (1.3 ml, 7.2 mmol) gegeben und das Gemisch wurde 30 min bei RT
gerührt. Anschließend wurde (+/-)-ie/f-Butyl-4-amino-3-(3,5-difluor-2-methoxyphenyl)-3-methyl- butanoat-Hydrochlorid (1.33 g, 95% Reinheit, 3.59 mmol, Beispiel 1 15A), gelöst in DMF (5 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethylacetat und Wasser (jeweils 20 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase zweimal mit Ethylacetat (jeweils 20 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/ Ethylacetat 85:15, Isolera One) gereinigt. Es wurden 988 mg (98% Reinheit, 63% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.64 min; MS (ESIpos): m/z = 639/641 [M+H]+
1H-NMR (600 MHz, DMSO-d6) δ [ppm]: 1.178 (16.00), 1.520 (2.02), 2.524 (0.70), 3.985 (2.10), 3.989 (2.04), 7.496 (0.44), 7.507 (0.63), 7.51 1 (0.70), 7.525 (1.1 1 ), 7.536 (0.69), 7.538 (0.75), 7.542 (0.96), 7.945 (0.70), 7.960 (0.52), 8.665 (0.40).
1 H-NMR (600 MHz, DMSO-d6): δ [ppm] = 8.67 (t, 1 H), 7.95 (d, 1 H), 7.86 (d, 1 H), 7.67 (br. s, 1 H), 7.57-7.48 (m, 5H), 7.30-7.22 (m, 1 H), 6.88 (d, 1 H), 4.10 (dd, 1 H), 3.99 (d, 3H), 3.64 (dd, 1 H), 3.16 (d, 1 H), 2.53 (verdeckt, 1 H), 2.33-1.97 (br., 3H), 1.52 (s, 3H), 1.18 (s, 9H).
Trennung der Enantiomere:
Die Titelverbindung (835 mg) wurde in einem Gemisch aus Isopropanol (25 ml) und Acetonitril (5 ml) aufgenommen, auf 50 °C erwärmt, filtriert und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 220A und 221A) [Säule: Daicel Chiralpak IC, 250 mm x 20 mm; Fluss: 100 ml/min; Detektion: 220 nm; Temperatur: 40 °C; Injektion: 1.0 ml; Eluent: 70% Kohlendioxid / 30% Ethanol; isokratisch, Laufzeit 4.0 min]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Beispiel 220A
(+)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(3,5-difluor-2- methoxyphenyl)-3-methylbutanoat (Enantiomer 1)
Bei der in Beispiel 219A beschriebenen Enantiomerentrennung wurden 300 mg (98% Reinheit, ee-Wert 93%) der Titelverbindung als früher eluierendes Enantiomer erhalten. [a]D 20 = +15.7°, 589 nm, c = 0.36 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.61 min; MS (ESIpos): m/z = 639/641 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.181 (16.00), 1.522 (2.09), 3.140 (0.40), 3.983 (2.49), 3.988 (2.48), 7.506 (0.89), 7.512 (0.70), 7.524 (1.38), 7.537 (1.67), 7.863 (0.49), 7.868 (0.47), 7.940 (0.97), 7.962 (0.62), 8.647 (0.41 ).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.65 (t, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.63 (br. s, 1 H), 7.57-7.48 (m, 5H), 7.31 -7.19 (m, 1 H), 6.88 (br. d, 1 H), 4.09 (dd, 1 H), 3.99 (d, 3H), 3.65 (dd, 1 H), 3.16 (d, 1 H), 2.53 (verdeckt, 1 H), 2.16 (br. s, 3H), 1.52 (s, 3H), 1.18 (s, 9H).
Beispiel 221A
(-)-ie f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]ami
methoxyphenyl)-3-methylbutanoat (Enantiomer 2)
Bei der in Beispiel 219A beschriebenen Enantiomerentrennung wurden 310 mg (98% Reinheit, ee-Wert 97%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = -15.6°, 589 nm, c = 0.34 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.61 min; MS (ESIpos): m/z = 639/641 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.180 (16.00), 1.521 (2.76), 3.140 (0.52), 3.175 (0.48), 3.983 (3.03), 3.988 (2.98), 6.865 (0.41 ), 6.891 (0.40), 7.492 (0.44), 7.506 (1.24), 7.524 (1.86), 7.537 (2.20), 7.841 (0.42), 7.863 (0.63), 7.939 (1.04), 7.962 (0.67), 8.646 (0.56).
1 H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.65 (t, 1 H), 7.95 (d, 1 H), 7.85 (dd, 1 H), 7.68 (br. s, 1 H), 7.58-7.46 (m, 5H), 7.31-7.19 (m, 1 H), 6.88 (d, 1 H), 4.09 (dd, 1 H), 3.99 (d, 3H), 3.65 (dd, 1 H), 3.16 (d, 1 H), 2.53 (verdeckt, 1 H), 2.16 (br. s, 3H), 1.52 (s, 3H), 1.18 (s, 9H).
Beispiel 222A
(+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(4-fluor-2-methoxy- phenyl)-3-methylbutanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (1.54 g, 4.50 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (15 ml) wurden HATU (2.56 g, 6.74 mmol) und DIPEA (2.3 ml, 13 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-4-amino-3-(4-fluor-2-methoxyphenyl)-3-methylbutanoat-
Hydrochlorid (2.37 g, 95% Reinheit, 6.74 mmol, Beispiel 116A), gelöst in DMF (9 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethylacetat und Wasser (jeweils 40 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase zweimal mit Ethylacetat (jeweils 40 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/ Ethylacetat 85:15, Isolera One) gereinigt. Es wurden 1.89 g (98% Reinheit, 66% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.59 min; MS (ESIpos): m/z = 621/623 [M+H]+
1H-NMR (600 MHz, DMSO-d6) δ [ppm]: 1.1 16 (16.00), 1.531 (1.67), 2.388 (0.59), 2.410 (0.60), 3.169 (0.48), 3.191 (0.48), 3.336 (6.51 ), 7.500 (0.53), 7.504 (0.72), 7.517 (1.18), 7.528 (1.46), 7.929 (0.79), 7.943 (0.57).
1H-NMR (600 MHz, DMSO-d6): δ [ppm] = 8.57 (t, 1 H), 7.94 (d, 1 H), 7.84 (dd, 1 H), 7.67 (br. s, 1 H), 7.56-7.47 (m, 5H), 7.18 (dd, 1 H), 6.90 (d, 1 H), 6.70 (br. t, 1 H), 4.24 (dd, 1 H), 3.87 (s, 3H), 3.57 (dd, 1 H), 3.18 (d, 1 H), 2.40 (d, 1 H), 2.10 (br. s, 3H), 1.53 (s, 3H), 1.12 (s, 9H).
Trennung der Enantiomere:
Die Titelverbindung (1.73 g) wurde in einem Gemisch aus Isopropanol (95 ml) und Acetonitril (5 ml) aufgenommen, auf 50 °C erwärmt, filtriert und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 223A und 224A) [Säule: Daicel Chiralpak IC, 250 mm x 20 mm; Fluss: 100 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 6.0 ml; Eluent: Kohlendioxid / Ethanol / Methanol 7:3:0— > 7:0:3 ; isokratisch, Laufzeit 14.0 min]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet.
Beispiel 223A
(+)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(4-fluor-2-methoxy- phenyl)-3-methylbutanoat (Enantiomer 1)
Bei der in Beispiel 222A beschriebenen Enantiomerentrennung wurden 750 mg (98% Reinheit, ee-Wert 100%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = +1 1.1 °, 589 nm, c = 0.40 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.36 min; MS (ESIpos): m/z = 621/623 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.030 (0.95), 1.045 (0.95), 1.120 (16.00), 1.531 (2.02), 2.388 (0.61 ), 2.421 (0.65), 3.159 (0.56), 3.192 (0.53), 3.869 (3.82), 7.175 (0.44), 7.178 (0.42), 7.498 (0.82), 7.506 (0.74), 7.517 (1.89), 7.522 (1.69), 7.849 (0.54), 7.854 (0.51 ), 7.923 (1.03), 7.946 (0.64).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.55 (t, 1 H), 7.93 (d, 1 H), 7.84 (dd, 1 H), 7.67 (br. s, 1 H), 7.55-7.47 (m, 5H), 7.18 (dd, 1 H), 6.89 (dd, 1 H), 6.70 (td, 1 H), 4.23 (dd, 1 H), 3.87 (s, 3H), 3.57 (dd, 1 H), 3.18 (d, 1 H), 2.41 (d, 1 H), 2.1 1 (br. s, 3H), 1.53 (s, 3H), 1.12 (s, 9H).
Beispiel 224A
(-)-ie f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(4-fl
phenyl)-3-methylbutanoat (Enantiomer 2)
Bei der in Beispiel 222A beschriebenen Enantiomerentrennung wurden 820 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = -1 1.5°, 589 nm, c = 0.38 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.36 min; MS (ESIpos): m/z = 621/623 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.030 (3.00), 1.046 (3.01 ), 1.120 (16.00), 1.531 (1.99), 2.389 (0.60), 2.422 (0.65), 3.160 (0.56), 3.193 (0.52), 3.869 (3.80), 7.175 (0.43), 7.179 (0.42), 7.498 (0.81 ), 7.506 (0.71 ), 7.517 (1.85), 7.523 (1.67), 7.849 (0.53), 7.854 (0.51 ), 7.923 (1.03), 7.946 (0.65).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.55 (t, 1 H), 7.93 (d, 1 H), 7.84 (dd, 1 H), 7.67 (br. s, 1 H), 7.56-7.46 (m, 5H), 7.18 (dd, 1 H), 6.89 (dd, 1 H), 6.70 (td, 1 H), 4.23 (dd, 1 H), 3.87 (s, 3H), 3.57 (dd, 1 H), 3.18 (d, 1 H), 2.41 (d, 1 H), 2.1 1 (br. s, 3H), 1.53 (s, 3H), 1.12 (s, 9H).
Beispiel 225A
(+/-)-Ethyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2,2-difluor-3-(2-methoxy- phenyl)butanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (95 mg, 278 μηηοΙ, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (1.5 ml) wurden HATU (137 mg, 361 μηηοΙ) und DIPEA (120 μΙ, 690 μηηοΙ) gegeben, und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-Ethyl-4-amino-2,2-difluor-3-(2-methoxyphenyl)butanoat-
Hydrochlorid (86 mg, 278 μηηοΙ, Beispiel 1 18A hinzugegeben, und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch mittels praparativer HPLC (Chromato- rex C18, 10 μΜ, 125 x 30 mm, Aceton itril/Wasser-Gradient mit 0.01 % TFA) gereinigt. Zwei getrennte Fraktionen wurden gesammelt, eingeengt und die beiden Rückstände lyophilisiert. Aus dem einen Rückstand wurden 46 mg (60% Reinheit, 17% d. Th., s. Analytik) der Titelverbindung erhalten. Aus dem anderen Rückstand wurden nach erneuter Reinigung mittels praparativer HPLC (Chromatorex C18, 10 μΜ, 125 x 30 mm, Acetonitril/Wasser-Gradient mit 0.01 % TFA) 8.8 mg (96% Reinheit, 5% d. Th.) der korrespondierenden Carbonsäure (+/-)-4-{[(6-Brom-3-methyl- 2-phenylchinolin-4-yl)carbonyl]amino}-2,2-difluor-3-(2-methoxyphenyl)butansäure erhalten.
LC-MS (Methode 7): Rt = 1.56 min; MS (ESIpos): m/z = 597/599 [M+H]+ Beispiel 226A
(+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-fluor-6-methoxy- phenyl)-3-methylbutanoat (Racemat)

Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (342 mg, 1.00 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (4 ml) wurden HATU (456 mg, 1.20 mmol) und DIPEA (420 μΙ, 2.4 mmol) gegeben und das Gemisch wurde 15 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-4-amino-3-(2-fluor-6-methoxyphenyl)-3-methyl- butanoat (363 mg, 82% Reinheit, 1.00 mmol, Beispiel 119A), gelöst in DMF (3 ml) hinzugege- ben, und das Gemisch wurde 3 h bei 60 °C und danach über Nacht bei RT gerührt. Anschließend wurde das Gemisch mit Ethylacetat und Wasser (jeweils 100 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (80 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung (100 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (50 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/ Ethylacetat 97:3 — > 7:3, Isolera One) gereinigt. Die
vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 288 mg (84% Reinheit, 39% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.57 min; MS (ESIpos): m/z = 621/623 [M+H]+
1H-NMR (500 MHz, DMSO-d6): δ [ppm] = 8.68 (t, 1 H), 7.94 (d, 1 H), 7.84 (dd, 1 H), 7.66 (br. s, 1 H), 7.55-7.47 (m, 5H), 7.25-7.18 (m, 1 H), 6.84 (d, 1 H), 6.68 (dd, 1 H), 4.24 (br. dd, 1 H), 3.84 (s, 3H), 3.57 (br. dd, 1 H), 3.34 (d, 1 H), 2.35 (d, 1 H), 2.12 (br. s, 3H), 1.69 (d, 3H), 1.17 (s, 9H).
Trennung der Enantiomere:
Die Titelverbindung (215 mg) wurde mittels präparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 227A und 228A) [Säule: Daicel Chiralpak ID, 5 μηη, 250 mm x 20 mm; Fluss: 15 ml/min; Detektion: 220 nm; Temperatur: 25 °C; Injektion: 0.15 ml; Eluent: 50% Heptan / 50% Ethanol; Laufzeit 10 min, isokratisch].
Beispiel 227A
(-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-fluor-6-methoxy- phenyl)-3-methylbutanoat (Enantiomer 1)
Bei der in Beispiel 226A beschriebenen Enantiomerentrennung wurden 76 mg (87% Reinheit, ee-Wert 97%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -8.0°, 436 nm, c = 0.34 g/100 ml, Chloroform
LC-MS (Methode 1 ): Rt = 2.57 min; MS (ESIpos): m/z = 621/623 [M+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.67 (t, 1 H), 7.94 (d, 1 H), 7.84 (dd, 1 H), 7.64 (br. s, 1 H), 7.55-7.47 (m, 5H), 7.26-7.17 (m, 1 H), 6.84 (d, 1 H), 6.68 (dd, 1 H), 4.24 (br. dd, 1 H), 3.84 (s, 3H), 3.57 (br. dd, 1 H), 3.34 (d, 1 H, teilweise verdeckt), 2.35 (d, 1 H), 2.1 1 (br. s, 3H), 1.69 (d, 3H), 1.17 (s, 9H).
Beispiel 228A
(+)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-fluor-6-methoxy- phenyl)-3-methylbutanoat (Enantiomer 2)
Bei der in Beispiel 226A beschriebenen Enantiomerentrennung wurden 61 mg (100% Reinheit, ee-Wert 94%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = +3.7°, 436 nm, c = 0.31 g/100 ml, Chloroform
LC-MS (Methode 1 ): Rt = 2.57 min; MS (ESIpos): m/z = 621/623 [M+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.68 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.64 (br. s, 1 H), 7.56-7.46 (m, 5H), 7.26-7.17 (m, 1 H), 6.84 (d, 1 H), 6.68 (dd, 1 H), 4.24 (dd, 1 H), 3.84 (s,
3H), 3.57 (dd, 1 H), 3.34 (d, 1 H, teilweise verdeckt), 2.35 (d, 1 H), 2.1 1 (br. s, 3H), 1.69 (d, 3H), 1.17 (s, 9H).
Beispiel 229A
ie f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amin
(Diastereomerengemisch)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (414 mg, 1.21 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (5 ml) wurden HATU (552 mg, 1.45 mmol) und DIPEA (510 μΙ, 2.9 mmol) gegeben und das Gemisch wurde 15 min bei RT gerührt. Anschließend wurde ie/f-Butyl-4-amino-3-(2-chlorphenyl)pentanoat (462 mg, 74% Reinheit, 1.21 mmol, Beispiel 120A), gelöst in DMF (3 ml) hinzugegeben, und das Gemisch wurde 22 h bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch direkt (ohne weitere Aufarbeitung) mittels präparativer HPLC an achiraler Phase (Methode 14) gereinigt. Die vereinigten Zielfraktionen wurden jeweils eingeengt, und die betreffenden Rückstände wurde im Vakuum getrocknet (siehe Beispiele 230A und 231 A).
Beispiel 230A
(+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-chlorphenyl)- pentanoat (Racemat 1)
Bei der in Beispiel 229A beschriebenen Diastereomerentrennung wurden 123 mg (100% Rein- heit) der Titelverbindung als früher eluierendes Racemat erhalten.
LC-MS (Methode 1 ): Rt = 2.61 min; MS (ESIpos): m/z = 607/609 [M+H]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.200 (16.00), 1.234 (1.15), 1.245 (1.94), 1.262 (1.83), 2.710 (0.58), 2.734 (0.56), 2.760 (0.63), 2.773 (0.67), 3.747 (0.40), 3.759 (0.42), 7.490 (0.69), 7.505 (2.14), 7.51 1 (1.76), 7.523 (3.09), 7.536 (1.81 ), 7.831 (0.42), 7.853 (0.59), 7.929 (0.98), 7.951 (0.63).
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.72 (br. s, 1 H), 7.94 (d, 1 H), 7.84 (d, 1 H), 7.78-7.15 (m, 10H), 4.72-4.57 (m, 1 H), 3.81 -3.69 (m, 1 H), 2.86-2.63 (m, 2H), 2.39-1.69 (br. m, 3H), 1.25 (d, 3H), 1.20 (s, 9H).
Beispiel 231A
(+/-)-ie f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]am
pentanoat (Racemat 2)
Bei der in Beispiel 229A beschriebenen Diastereomerentrennung wurden 148 mg (75% Reinheit) einer ersten Charge, 56 mg (89% Reinheit, siehe Analytik) einer zweiten Charge und 57 mg (40% Reinheit) einer dritten Charge der Titelverbindung als später eluierendes Racemat erhal- ten. Als Nebenkomponente war jeweils die Verbindung aus Beispiel 230A (Racemat 1) zu unterschiedlichen Anteilen enthalten.
LC-MS (Methode 1 ): Rt = 2.66 min; MS (ESIpos): m/z = 607/609 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.89 (br. s, 1 H), 7.99 (d, 1 H), 7.89 (dd, 1 H), 7.74 (br. s, 1 H), 7.72-7.24 (m, 10H), 4.52-4.39 (m, 1 H), 3.82-3.71 (m, 1 H), 2.95-2.75 (m, 2H), 2.30 (br. s, 3H), 1.21 (d, 3H), 1.15 (s, 9H).
Beispiel 232A
(+/-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlorphenyl)- pentanoat (Racemat)

Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (1.16 g, 3.38 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (15 ml) wurden HATU (1.93 g, 5.07 mmol) und DIPEA (1.8 ml, 10 mmol) gegeben, und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-5-amino-4-(2-chlorphenyl)pentanoat (1.58 g, 91% Reinheit, 5.07 mmol, Beispiel 121A), gelöst in DMF (5 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethylacetat und Wasser
(jeweils 30 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase zweimal mit Ethylacetat (jeweils 30 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulen- Chromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/ Ethylacetat 85:15, Isolera One) vorgereinigt. Anschließend wurde mittels präparativer HPLC (Methode 16) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 1.43 g (98% Reinheit, 68% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.63 min; MS (ESIpos): m/z = 607/609 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.152 (0.02), 1.206 (0.07), 1.290 (0.09), 1.31 1 (0.17), 1.334 (0.13), 1.367 (16.00), 1.393 (0.24), 1.523 (0.07), 1.772 (0.06), 1.804 (0.16), 1.823 (0.21 ), 1.846 (0.12), 2.01 1 (0.07), 2.031 (0.17), 2.043 (0.19), 2.060 (0.23), 2.082 (1.29), 2.094 (0.71 ), 2.1 1 1 (0.40), 2.1 19 (0.45), 2.139 (0.78), 2.253 (0.04), 2.327 (0.03), 2.366 (0.02), 2.670 (0.03), 2.710 (0.02), 3.61 1 (0.23), 3.718 (0.32), 7.246 (0.17), 7.264 (0.37), 7.283 (0.26), 7.354 (0.23), 7.373 (0.41 ), 7.391 (0.21 ), 7.436 (0.63), 7.456 (0.51 ), 7.499 (1.28), 7.507 (0.85), 7.518 (3.1 1 ), 7.696 (0.09), 7.830 (0.40), 7.835 (0.37), 7.853 (0.61 ), 7.858 (0.59), 7.928 (1.12), 7.950 (0.71 ), 8.837 (0.24), 8.851 (0.47), 8.865 (0.23).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.85 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.68 (br. s, 1 H), 7.55-7.48 (m, 6H), 7.45 (d, 1 H), 7.37 (br. t, 1 H), 7.27 (br. t, 1 H), 3.81-3.53 (m, 3H), 2.20-1.98 (m, 6H), 1.89-1.75 (m, 1 H), 1.37 (s, 9H).
Beispiel 233A
(+/-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlorphenyl)-4- methylpentanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (470 mg, 1.37 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (6.0 ml) wurden HATU (783 mg, 2.06 mmol) und DIPEA (720 μΙ, 4.1 mmol) gegeben und das Gemisch wurde 30 min bei RT
gerührt. Anschließend wurde (+/-)-ie/f-Butyl-5-amino-4-(2-chlorphenyl)-4-methylpentanoat (840 mg, 73% Reinheit, 2.06 mmol, Beispiel 122A), gelöst in DMF (2 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethylacetat und Wasser (jeweils 30 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase zweimal mit Ethylacetat (30 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/ Ethylacetat 85:15, Isolera One) vorgereinigt. Anschließend wurde mittels präparativer HPLC (Me- thode 16) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 309 mg (98% Reinheit, 35% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.70 min; MS (ESIpos): m/z = 621/623 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.188 (0.07), 1.233 (0.04), 1.280 (0.14), 1.297 (0.08), 1.348 (16.00), 1.375 (0.18), 1.512 (2.18), 1.717 (0.1 1 ), 1.729 (0.13), 1.745 (0.17), 1.756 (0.28), 1.767 (0.15), 1.783 (0.21 ), 1.796 (0.20), 1.872 (0.19), 1.885 (0.21 ), 1.907 (0.24), 1.919 (0.23), 1.934 (0.12), 1.947 (0.19), 1.986 (0.23), 1.999 (0.19), 2.015 (0.27), 2.026 (0.35), 2.037 (0.22), 2.053 (0.26), 2.065 (0.24), 2.317 (0.06), 2.365 (0.04), 2.634 (0.12), 2.647 (0.13), 2.668 (0.22), 2.696 (0.12), 2.708 (0.12), 3.666 (0.28), 3.679 (0.31 ), 3.699 (0.33), 3.713 (0.31 ), 3.922 (0.27), 4.339 (0.22), 4.357 (0.24), 4.373 (0.22), 4.390 (0.20), 7.257 (0.12), 7.275 (0.32), 7.298 (0.40), 7.320 (0.41 ), 7.337 (0.21 ), 7.356 (0.06), 7.375 (0.04), 7.408 (0.57), 7.425 (0.69), 7.445 (0.29), 7.473 (0.1 1 ), 7.498 (0.83), 7.506 (0.76), 7.517 (2.01 ), 7.668 (0.07), 7.828 (0.36), 7.833 (0.33), 7.850 (0.55), 7.855 (0.52), 7.924 (1.00), 7.946 (0.64), 8.614 (0.25), 8.629 (0.41 ), 8.644 (0.25).
Trennung der Enantiomere:
Die Titelverbindung (220 mg) wurde in einem Gemisch aus Ethanol (7 ml) und Acetonitril (3 ml) in der Wärme gelöst und mittels präparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 234A und 235A) [Säule: Daicel Chiralpak IC, 5 μηη, 250 mm x 20 mm; Fluss: 15 ml/min; Detektion: 210 nm; Temperatur: 25 °C; Injektion: 0.5 ml; Eluent: 20% Ethanol / 80% Heptan; Laufzeit 20 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert. Beispiel 234A
(-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlorphenyl)-4- methylpentanoat (Enantiomer 1)
Bei der in Beispiel 233A beschriebenen Enantiomerentrennung wurden 91 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als früher eluierendes Enantiomer erhalten. [a]D 20 = -33.9°, 589 nm, c = 0.25 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.68 min; MS (ESIpos): m/z = 621/623 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.007 (0.49), 1.280 (0.15), 1.348 (16.00), 1.512 (1.93), 1.728 (0.12), 1.756 (0.24), 1.783 (0.19), 1.795 (0.18), 1.872 (0.16), 1.884 (0.17), 1.919 (0.20), 1.946 (0.16), 1.986 (0.20), 2.026 (0.29), 2.053 (0.22), 2.669 (0.23), 2.708 (0.13), 3.665 (0.23), 3.679 (0.25), 3.699 (0.27), 3.713 (0.25), 4.338 (0.19), 4.356 (0.20), 4.372 (0.19), 4.389 (0.18), 7.276 (0.28), 7.298 (0.36), 7.322 (0.36), 7.337 (0.17), 7.405 (0.48), 7.428 (0.60), 7.446 (0.27), 7.497 (0.74), 7.505 (0.68), 7.516 (1.81 ), 7.826 (0.34), 7.831 (0.30), 7.848 (0.52), 7.853 (0.49), 7.922 (1.01 ), 7.945 (0.63), 8.612 (0.22), 8.627 (0.35), 8.643 (0.21 ).
Beispiel 235A
(+)-ie f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-ch
methylpentanoat (Enantiomer 2)
Bei der in Beispiel 233A beschriebenen Enantiomerentrennung wurden 96 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = +35.1 °, 589 nm, c = 0.26 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.68 min; MS (ESIpos): m/z = 621/623 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.007 (0.35), 1.188 (0.07), 1.280 (0.15), 1.297 (0.08), 1.348 (16.00), 1.512 (2.03), 1.717 (0.10), 1.728 (0.12), 1.745 (0.16), 1.756 (0.26), 1.783 (0.20), 1.795 (0.19), 1.872 (0.16), 1.885 (0.19), 1.919 (0.22), 1.947 (0.17), 1.986 (0.21 ), 1.999 (0.17), 2.015 (0.24), 2.025 (0.30), 2.053 (0.24), 2.065 (0.21 ), 2.327 (0.07), 2.669 (0.22), 2.709 (0.13), 3.665 (0.24), 3.679 (0.26), 3.699 (0.29), 3.712 (0.27), 4.338 (0.20), 4.356 (0.22), 4.372 (0.20), 4.389 (0.18), 7.275 (0.30), 7.298 (0.38), 7.319 (0.38), 7.337 (0.18), 7.405 (0.51 ), 7.424 (0.63), 7.445 (0.28), 7.497 (0.77), 7.505 (0.71 ), 7.516 (1.87), 7.826 (0.34), 7.831 (0.31 ), 7.848 (0.52), 7.853 (0.49), 7.922 (1.02), 7.944 (0.64), 8.612 (0.23), 8.627 (0.37), 8.642 (0.22).
Beispiel 236A
(+/-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlor-6-fluor- phenyl)pentanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (608 mg, 1.78 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (8.0 ml) wurden HATU (1.01 g, 2.66 mmol) und DIPEA (930 μΙ, 5.3 mmol) gegeben und das Gemisch wurde 30 min bei RT ge- rührt. Anschließend wurde (+/-)-ierf-Butyl-5-amino-4-(2-chlor-6-fluorphenyl)pentanoat (1.20 g, 67% Reinheit, 2.66 mmol, Beispiel 123A), gelöst in DMF (2 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethylacetat und Wasser (jeweils 20 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase zweimal mit Ethylacetat (20 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesät- tigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat 85:15, Isolera One) vorgereinigt. Anschließend wurde mittels präparativer HPLC (Methode 16) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 604 mg (98% Reinheit, 53% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.60 min; MS (ESIpos): m/z = 625/627 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.361 (16.00), 2.1 19 (0.88), 2.135 (0.88), 2.158 (1.67), 7.330 (0.95), 7.501 (0.94), 7.509 (0.86), 7.521 (3.1 1 ), 7.838 (0.48), 7.843 (0.44), 7.860 (0.72), 7.865 (0.69), 7.933 (1.26), 7.955 (0.78), 8.938 (0.47).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.94 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.72 (br. s, 1 H), 7.56-7.45 (m, 5H), 7.37-7.28 (m, 2H), 7.27-7.16 (m, 1 H), 3.89-3.53 (m, 3H), 2.21-1.90 (m, 7H), 1.36 (s, 9H).
Beispiel 237A
(+/-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-methylphenyl)- pentanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (400 mg, 1.17 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (4.0 ml) wurden HATU (667 mg, 1.75 mmol) und DIPEA (610 μΙ, 3.5 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ierf-Butyl-5-amino-4-(2-methylphenyl)pentanoat (486 mg, 95% Reinheit, 1.75 mmol, Beispiel 124A), gelöst in DMF (2.5 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethylacetat und Wasser (jeweils 20 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase zweimal mit Ethylacetat (20 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesät- tigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat 85:15, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert. Es wurden 429 mg (98% Reinheit, 61% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.61 min; MS (ESIpos): m/z = 587/589 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.009 (0.21 ), 0.007 (0.23), 1.202 (0.07), 1.304 (0.08), 1.336 (0.13), 1.362 (16.00), 1.397 (0.14), 1.518 (0.06), 1.747 (0.05), 1.779 (0.15), 1.804 (0.19), 1.821 (0.13), 2.029 (0.20), 2.041 (0.22), 2.060 (1.22), 2.072 (0.80), 2.093 (0.46), 2.109 (0.40), 2.328 (3.10), 2.365 (0.05), 2.669 (0.05), 2.709 (0.04), 3.483 (0.10), 3.496 (0.14), 3.515 (0.18), 3.530 (0.18), 3.545 (0.1 1 ), 3.698 (0.14), 3.716 (0.22), 3.734 (0.20), 3.749 (0.17), 3.768 (0.10), 7.094 (0.13), 7.1 12 (0.34), 7.130 (0.30), 7.165 (0.56), 7.183 (0.32), 7.21 1 (0.34), 7.229 (0.18), 7.301 (0.55), 7.321 (0.38), 7.490 (0.23), 7.500 (0.69), 7.508 (0.67), 7.519 (2.17), 7.530 (0.94), 7.694 (0.12), 7.835 (0.37), 7.840 (0.34), 7.857 (0.56), 7.863 (0.55), 7.933 (1.02), 7.955 (0.65), 8.820 (0.21 ), 8.835 (0.34), 8.848 (0.21 ).
Trennung der Enantiomere:
Die Titelverbindung (390 mg) wurde in Isopropanol (4 ml) gelöst und mittels präparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 238A und 239A) [Säule: Daicel
Chiralpak AD-H, 5 μηι, 250 mm x 20 mm; Fluss: 20 ml/min; Detektion: 210 nm; Temperatur: 23 °C; Injektion: 0.25 ml; Eluent: 20% Isopropanol / 80% Heptan; Laufzeit 10 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert.
Beispiel 238A
(-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-methylphenyl)- pentanoat (Enantiomer 1)
Bei der in Beispiel 237A beschriebenen Enantiomerentrennung wurden 128 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -1 1.1 °, 589 nm, c = 0.35 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.63 min; MS (ESIpos): m/z = 587/589 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.201 (0.07), 1.304 (0.09), 1.336 (0.15), 1.362 (16.00), 1.518 (0.06), 1.748 (0.06), 1.779 (0.16), 1.804 (0.20), 1.822 (0.14), 1.853 (0.04), 2.014 (0.16), 2.029 (0.22), 2.041 (0.25), 2.060 (1.31 ), 2.072 (0.85), 2.093 (0.49), 2.108 (0.42), 2.328 (3.28), 2.365 (0.04), 2.669 (0.03), 2.709 (0.03), 3.304 (0.19), 3.321 (0.20), 3.483 (0.12), 3.496 (0.16), 3.515 (0.20), 3.530 (0.20), 3.545 (0.13), 3.698 (0.18), 3.716 (0.28), 3.734 (0.25), 3.750 (0.23), 3.768 (0.17), 3.876 (0.18), 7.093 (0.14), 7.1 1 1 (0.36), 7.130 (0.32), 7.164 (0.60), 7.182 (0.34), 7.21 1 (0.36), 7.229 (0.19), 7.301 (0.59), 7.320 (0.41 ), 7.490 (0.25), 7.501 (0.75), 7.508 (0.71 ), 7.520 (2.27), 7.531 (0.97), 7.695 (0.13), 7.836 (0.38), 7.841 (0.34), 7.859 (0.59), 7.864 (0.56), 7.934 (1.07), 7.956 (0.68), 8.821 (0.23), 8.836 (0.36), 8.850 (0.22). Beispiel 239A
(+)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-methylphenyl)- pentanoat (Enantiomer 2)
Bei der in Beispiel 237A beschriebenen Enantiomerentrennung wurden 128 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als später eluierendes Enantiomer erhalten. [a]D 20 = +16.8°, 589 nm, c = 0.37 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.40 min; MS (ESIpos): m/z = 587/589 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.009 (0.32), 0.008 (0.27), 1.078 (0.08), 1.093 (0.08), 1.202 (0.06), 1.261 (0.17), 1.304 (0.08), 1.336 (0.13), 1.362 (16.00), 1.518 (0.06), 1.779 (0.14), 1.804 (0.18), 2.029 (0.18), 2.060 (1.12), 2.072 (0.73), 2.094 (0.42), 2.328 (2.92), 3.496 (0.13), 3.515 (0.16), 3.530 (0.17), 3.697 (0.14), 3.716 (0.21 ), 3.733 (0.18), 3.749 (0.16), 3.768 (0.09), 7.093 (0.12), 7.1 12 (0.31 ), 7.130 (0.28), 7.165 (0.53), 7.182 (0.29), 7.21 1 (0.31 ), 7.229 (0.17), 7.302 (0.51 ), 7.320 (0.35), 7.490 (0.22), 7.500 (0.67), 7.508 (0.64), 7.519 (2.04), 7.531 (0.90),
7.695 (0.12), 7.835 (0.38), 7.840 (0.34), 7.857 (0.57), 7.863 (0.55), 7.933 (1.05), 7.955 (0.66), 8.820 (0.19), 8.835 (0.31 ), 8.849 (0.19).
Beispiel 240A
(+/-)-ie f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4- phenyl)pentanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (3.17 g, 9.28 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (30 ml) wurden HATU (5.29 g, 13.9 mmol) und DIPEA (4.8 ml, 28 mmol) gegeben und das Gemisch wurde 30 min bei RT ge- rührt. Anschließend wurde (+/-)-ierf-Butyl-5-amino-4-(2-chlor-5-fluorphenyl)pentanoat (7.00 g, 60% Reinheit, 13.9 mmol, Beispiel 125A), gelöst in DMF (20 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethylacetat und Wasser (jeweils 150 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase zweimal mit Ethylacetat (150 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge Ultra, Cyclohexan/Ethylacetat 85:15, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert. Es wurden 4.03 g (91 % Reinheit, 63% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.61 min; MS (ESIpos): m/z = 625/627 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.22), 0.008 (0.21 ), 0.018 (0.08), 0.838 (0.09), 0.863 (0.04), 1.21 1 (0.07), 1.283 (0.08), 1.313 (0.1 1 ), 1.344 (4.94), 1.359 (0.72), 1.371 (16.00), 1.397 (0.20), 1.499 (0.04), 1.528 (0.07), 1.663 (0.06), 1.785 (0.14), 1.801 (0.16), 1.820 (0.18), 1.842 (0.14), 1.928 (0.06), 1.947 (0.07), 1.971 (0.19), 1.988 (0.26), 2.009 (0.25), 2.022 (0.23), 2.041 (0.26), 2.059 (0.24), 2.074 (0.21 ), 2.093 (0.72), 2.1 10 (0.92), 2.139 (1.05), 2.328 (0.05), 2.366 (0.05), 2.690 (0.17), 2.710 (0.12), 3.223 (0.06), 3.607 (0.19), 3.735 (0.37), 7.035 (0.07), 7.056 (0.12), 7.077 (0.09), 7.116 (0.18), 7.140 (0.30), 7.155 (0.21 ), 7.181 (0.08), 7.188 (0.07),
7.394 (0.10), 7.41 1 (0.36), 7.418 (0.38), 7.436 (0.38), 7.444 (0.32), 7.478 (0.47), 7.491 (0.59), 7.500 (1 .12), 7.508 (0.83), 7.518 (2.77), 7.530 (1 .02), 7.664 (0.06), 7.832 (0.42), 7.837 (0.37), 7.854 (0.64), 7.859 (0.60), 7.929 (1 .10), 7.951 (0.71 ), 8.842 (0.20), 8.856 (0.39), 8.870 (0.19).
Trennung der Enantiomere:
Die Titelverbindung (4.00 g) wurde in Ethanol (40 ml) gelöst und mittels praparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 241A und 242A) [Säule: Daicel Chi- ralcel OX-H, 5 [Jim , 250 mm x 20 mm; Fluss: 40 ml/min; Injektion: 0.13 ml; Eluent: 20% Ethanol / 80% Heptan; Laufzeit 9 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert.
Beispiel 241 A
(-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlor-5-fluor- phenyl)pentanoat (Enantiomer 1)
Bei der in Beispiel 240A beschriebenen Enantiomerentrennung wurden 1.57 g (86% Reinheit, ee-Wert 99%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -15.8°, 589 nm, c = 0.34 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.60 min; MS (ESIpos): m/z = 625/627 [M+H]+
1 H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1 .312 (0.15), 1 .344 (1 .32), 1 .370 (16.00), 1 .819 (0.18), 2.021 (0.25), 2.043 (0.26), 2.059 (0.27), 2.073 (0.28), 2.092 (0.75), 2.109 (0.94), 2.139 (1 .1 1 ), 3.607 (0.20), 3.734 (0.39), 7.136 (0.32), 7.410 (0.34), 7.417 (0.34), 7.435 (0.33), 7.477 (0.49), 7.490 (0.63), 7.499 (1 .20), 7.507 (0.89), 7.517 (3.06), 7.831 (0.45), 7.836 (0.40), 7.853 (0.69), 7.858 (0.64), 7.928 (1 .16), 7.951 (0.73), 8.856 (0.39).
Beispiel 242A
(+)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlor-5-fluor- phenyl)pentanoat (Enantiomer 2)
Bei der in Beispiel 240A beschriebenen Enantiomerentrennung wurden 1.28 g (98% Reinheit, ee-Wert 95%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = +15.1 °, 589 nm, c = 0.36 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.60 min; MS (ESIpos): m/z = 625/627 [M+H]+
1 H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.009 (0.20), 0.007 (0.19), 0.857 (0.07), 1 .027 (0.03), 1 .044 (0.04), 1 .086 (0.04), 1 .101 (0.04), 1 .210 (0.07), 1 .282 (0.09), 1 .331 (0.14), 1 .344 (1.33), 1 .370 (16.00), 1 .526 (0.07), 1 .784 (0.13), 1.800 (0.15), 1 .818 (0.17), 1 .841 (0.13), 2.008 (0.20), 2.021 (0.22), 2.039 (0.22), 2.059 (0.19), 2.073 (0.16), 2.092 (0.70), 2.109 (0.92), 2.138 (1 .04),
2.327 (0.05), 2.365 (0.04), 2.669 (0.05), 2.709 (0.05), 3.607 (0.18), 3.733 (0.37), 7.136 (0.24), 7.409 (0.33), 7.417 (0.34), 7.435 (0.34), 7.442 (0.32), 7.477 (0.47), 7.490 (0.59), 7.499 (1.1 1 ), 7.507 (0.79), 7.517 (2.61 ), 7.529 (0.96), 7.532 (0.87), 7.664 (0.06), 7.831 (0.42), 7.836 (0.38), 7.853 (0.64), 7.858 (0.61 ), 7.928 (1.12), 7.950 (0.71 ), 8.841 (0.20), 8.855 (0.39), 8.869 (0.19). Beispiel 243A
(+/-)-ie f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]am
pentanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (185 mg, 541 μηηοΙ, dar- stellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (3 ml) wurden HATU (308 mg, 811 μηηοΙ) und DIPEA (280 μΙ, 1.6 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ierf-Butyl-5-amino-4-(2-methoxyphenyl)pentanoat (267 mg, 85% Reinheit, 81 1 μηηοΙ, Beispiel 126A), gelöst in DMF (1.5 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethylacetat und Wasser (jeweils 20 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase zweimal mit Ethylacetat (20 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat 85:15, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert. Es wurden 213 mg (98% Reinheit, 64% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.57 min; MS (ESIpos): m/z = 603/605 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.009 (0.16), 0.007 (0.13), 1.200 (0.07), 1.300 (0.10), 1.325 (0.17), 1.360 (16.00), 1.516 (0.06), 1.788 (0.12), 1.806 (0.16), 1.841 (0.08), 1.948 (0.06), 1.969 (0.15), 1.981 (0.17), 1.988 (0.17), 1.999 (0.19), 2.004 (0.19), 2.028 (0.97), 2.044 (0.74), 2.062 (0.26), 2.143 (0.93), 2.263 (0.03), 2.327 (0.03), 2.365 (0.02), 2.669 (0.03), 2.709 (0.02), 3.233 (0.02), 3.393 (0.17), 3.591 (0.02), 3.618 (0.08), 3.633 (0.14), 3.651 (0.25), 3.666 (0.31 ),
3.688 (0.23), 3.704 (0.25), 3.724 (0.23), 3.737 (0.12), 3.749 (0.18), 3.773 (4.01 ), 6.922 (0.24), 6.941 (0.52), 6.959 (0.32), 6.976 (0.54), 6.997 (0.63), 7.197 (0.27), 7.217 (0.50), 7.223 (0.64), 7.241 (0.53), 7.488 (0.24), 7.498 (0.72), 7.507 (0.67), 7.518 (2.34), 7.529 (0.96), 7.714 (0.21 ), 7.829 (0.39), 7.834 (0.34), 7.852 (0.59), 7.857 (0.55), 7.926 (1.07), 7.948 (0.68), 8.752 (0.22), 8.767 (0.44), 8.781 (0.22).
Trennung der Enantiomere:
Die Titelverbindung (160 mg) wurde in Isopropanol (3 ml) gelöst und mittels praparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 244A und 245A) [Säule: Daicel Chiralcel AD-H, 5 μηι, 250 mm x 20 mm; Fluss: 25 ml/min; Injektion: 0.10 ml; Eluent: 40% Isop- ropanol / 60% Heptan; Laufzeit 14 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert.
Beispiel 244A
(-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-methoxyphenyl)- pentanoat (Enantiomer 1)
Bei der in Beispiel 243A beschriebenen Enantiomerentrennung wurden 63 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -9.6°, 589 nm, c = 0.32 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.57 min; MS (ESIpos): m/z = 603/605 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.154 (0.03), 0.141 (0.03), 1.100 (0.05), 1.1 16 (0.05), 1.195 (0.06), 1.296 (0.1 1 ), 1.321 (0.16), 1.356 (16.00), 1.512 (0.06), 1.801 (0.16), 1.964 (0.14), 1.976 (0.16), 1.994 (0.18), 2.023 (0.95), 2.039 (0.73), 2.057 (0.25), 2.138 (0.92), 2.258 (0.03), 2.322 (0.04), 2.361 (0.05), 2.664 (0.05), 2.705 (0.05), 3.614 (0.08), 3.629 (0.14), 3.646 (0.24), 3.662 (0.31 ), 3.683 (0.22), 3.699 (0.25), 3.719 (0.23), 3.732 (0.12), 3.769 (4.00), 6.918 (0.24), 6.936 (0.52), 6.955 (0.30), 6.972 (0.54), 6.992 (0.62), 7.193 (0.27), 7.213 (0.50), 7.218 (0.64), 7.236 (0.53), 7.484 (0.22), 7.494 (0.69), 7.502 (0.66), 7.514 (2.37), 7.525 (0.97), 7.710 (0.21 ), 7.826 (0.38), 7.831 (0.34), 7.848 (0.59), 7.853 (0.56), 7.922 (1.07), 7.944 (0.68), 8.748 (0.22), 8.762 (0.43), 8.777 (0.21 ).
Beispiel 245A
(+)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-methoxyphenyl)- pentanoat (Enantiomer 2)
Bei der in Beispiel 243A beschriebenen Enantiomerentrennung wurden 59 mg (98% Reinheit, ee-Wert 98%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = +1 1.3°, 589 nm, c = 0.36 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.57 min; MS (ESIpos): m/z = 603/605 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.029 (0.07), 1.045 (0.07), 1.105 (0.15), 1.120 (0.15), 1.200 (0.07), 1.300 (0.10), 1.325 (0.15), 1.360 (16.00), 1.516 (0.07), 1.805 (0.17), 1.968 (0.15), 1.980 (0.18), 1.999 (0.19), 2.028 (1.00), 2.044 (0.78), 2.062 (0.27), 2.143 (1.00), 2.327 (0.06), 2.668 (0.05), 3.394 (0.20), 3.618 (0.13), 3.633 (0.18), 3.651 (0.28), 3.667 (0.35), 3.688 (0.25), 3.704 (0.27), 3.724 (0.25), 3.738 (0.14), 3.773 (4.12), 6.923 (0.25), 6.941 (0.56), 6.959 (0.33), 6.977 (0.58), 6.997 (0.67), 7.197 (0.29), 7.222 (0.67), 7.241 (0.56), 7.489 (0.24), 7.500 (0.73), 7.508 (0.69), 7.520 (2.58), 7.716 (0.24), 7.831 (0.39), 7.837 (0.36), 7.854 (0.60), 7.859 (0.59), 7.927 (1.11 ), 7.949 (0.70), 8.753 (0.23), 8.768 (0.46), 8.782 (0.23). Beispiel 246A
(+/-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlor-5-fluor- phenyl)-4-methylpentanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (734 mg, 2.15 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (8 ml) wurden HATU (1.22 g, 3.22 mmol) und DIPEA (1.1 ml, 6.4 mmol) gegeben, und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ierf-Butyl-5-amino-4-(2-chlor-5-fluorphenyl)-4-methylpentanoat (1.54 g, 66% Reinheit, 3.22 mmol, Beispiel 127A), gelöst in DMF (4 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethylacetat und Wasser (jeweils 20 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase zweimal mit Ethylacetat (20 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/ Ethylacetat 85:15, Isolera One) vorgereinigt. Anschließend wurde mittels präparativer HPLC (Methode 16) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 377 mg (98% Reinheit, 27% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.69 min; MS (ESIpos): m/z = 639 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.151 (0.01 ), 1.192 (0.07), 1.280 (0.14), 1.306 (0.08), 1.352 (16.00), 1.500 (2.15), 1.740 (0.1 1 ), 1.753 (0.13), 1.768 (0.17), 1.780 (0.27), 1.791 (0.14), 1.807 (0.22), 1.819 (0.21 ), 1.874 (0.19), 1.885 (0.20), 1.908 (0.23), 1.914 (0.22), 1.919 (0.23), 1.936 (0.13), 1.948 (0.17), 2.029 (0.24), 2.042 (0.24), 2.058 (0.29), 2.069 (0.37), 2.080 (0.23), 2.096 (0.26), 2.108 (0.23), 2.324 (0.06), 2.365 (0.03), 2.618 (0.1 1 ), 2.630 (0.12), 2.659 (0.18), 2.680 (0.1 1 ), 2.692 (0.09), 3.634 (0.20), 3.647 (0.21 ), 3.668 (0.23), 3.680 (0.21 ), 4.384 (0.18), 4.401 (0.20), 4.417 (0.19), 4.434 (0.16), 4.863 (0.04), 7.148 (0.15), 7.176 (0.57), 7.183 (0.42), 7.204 (0.43), 7.21 1 (0.30), 7.248 (0.02), 7.489 (0.48), 7.503 (0.99), 7.509 (0.84), 7.521 (1.70), 7.530 (1.54), 7.833 (0.32), 7.838 (0.30), 7.856 (0.49), 7.860 (0.47), 7.931 (0.97), 7.953 (0.62), 8.629 (0.23), 8.644 (0.37), 8.658 (0.23).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.65 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.63 (br. s, 1 H),
7.56- 7.46 (m, 6H), 7.23-7.13 (m, 2H), 4.41 (dd, 1 H), 3.66 (dd, 1 H), 2.73-2.59 (m, 1 H), 2.15 (br. s, 3H), 2.13-2.01 (m, 1 H), 1.97-1.86 (m, 1 H), 1.84-1.72 (m, 1 H), 1.50 (s, 3H), 1.35 (s, 9H).
Trennung der Enantiomere:
Die Titelverbindung (330 mg) wurde in Methanol (25 ml) gelöst und mittels praparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 247A und 248A) [Säule: Daicel Chiral- pak AD-H, 5 μηη, 250 mm x 20 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 0.5 ml; Eluent: 88% Kohlendioxid / 12% Methanol; Laufzeit 18 min, isokratisch]. Die vereinig- ten Zielfraktionen wurden eingeengt, und der Rückstand wurde jeweils im Vakuum getrocknet.
Beispiel 247A
(-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlor-5-fluor- phenyl)-4-methylpentanoat (Enantiomer 1)
Bei der in Beispiel 246A beschriebenen Enantiomerentrennung wurden 146 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -29.3°, 589 nm, c = 0.34 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.69 min; MS (ESIpos): m/z = 639/671 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.352 (16.00), 1.499 (2.16), 1.778 (0.27), 1.805 (0.23), 1.884 (0.21 ), 1.906 (0.23), 2.068 (0.37), 2.655 (0.19), 3.644 (0.21 ), 3.665 (0.23), 4.399 (0.19), 7.175 (0.59), 7.203 (0.43), 7.501 (1.00), 7.508 (0.90), 7.520 (1.86), 7.831 (0.33), 7.853 (0.51 ), 7.929 (0.99), 7.951 (0.63), 8.641 (0.36).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.64 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.68 (br. s, 1 H),
7.57- 7.45 (m, 6H), 7.24-7.13 (m, 2H), 4.41 (dd, 1 H), 3.66 (dd, 1 H), 2.71 -2.60 (m, 1 H), 2.15 (br. s, 3H), 2.12-2.01 (m, 1 H), 1.97-1.86 (m, 1 H), 1.83-1.73 (m, 1 H), 1.50 (s, 3H), 1.35 (s, 9H).
Beispiel 248A
(+)-ie f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2
phenyl)-4-methylpentanoat (Enantiomer 2)
Bei der in Beispiel 246A beschriebenen Enantiomerentrennung wurden 139 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = +26.7°, 589 nm, c = 0.32 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.69 min; MS (ESIpos): m/z = 639/641 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.192 (0.07), 1.233 (0.06), 1.280 (0.16), 1.307 (0.07),
1.352 (16.00), 1.499 (1.97), 1.738 (0.10), 1.750 (0.12), 1.766 (0.16), 1.778 (0.25), 1.789 (0.13), 1.805 (0.21 ), 1.817 (0.20), 1.871 (0.17), 1.883 (0.19), 1.905 (0.21 ), 1.918 (0.21 ), 1.933 (0.12),
1.946 (0.16), 2.028 (0.23), 2.040 (0.22), 2.057 (0.26), 2.068 (0.33), 2.095 (0.24), 2.107 (0.21 ),
2.322 (0.09), 2.365 (0.05), 2.627 (0.12), 2.659 (0.17), 2.709 (0.05), 3.632 (0.18), 3.644 (0.19),
3.665 (0.21 ), 3.678 (0.19), 4.382 (0.16), 4.399 (0.17), 4.415 (0.16), 4.432 (0.14), 7.175 (0.53),
7.203 (0.40), 7.210 (0.27), 7.487 (0.43), 7.501 (0.88), 7.508 (0.79), 7.519 (1.62), 7.527 (1.39), 7.831 (0.30), 7.836 (0.27), 7.853 (0.45), 7.858 (0.43), 7.929 (0.89), 7.951 (0.57), 8.625 (0.20), 8.640 (0.33), 8.655 (0.20).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.64 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.66 (br. s, 1 H), 7.57-7.44 (m, 6H), 7.23-7.13 (m, 2H), 4.41 (dd, 1 H), 3.66 (dd, 1 H), 2.72-2.59 (m, 1 H), 2.15 (br. s, 3H), 2.13-2.02 (m, 1 H), 1.96-1.86 (m, 1 H), 1.84-1.72 (m, 1 H), 1.50 (s, 3H), 1.35 (s, 9H). Beispiel 249A
(+/-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlor-3,6-difluor- phenyl)pentanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (963 mg, 2.81 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (10 ml) wurden HATU (1.28
g, 3.38 mmol) und DIPEA (1.2 ml, 6.8 mmol) gegeben und das Gemisch wurde 15 min bei RT gerührt. Anschließend wurde (+/-)-ierf-Butyl-5-amino-4-(2-chlor-3,6-difluorphenyl)pentanoat (900 mg, 2.81 mmol, Beispiel 128A), gelöst in DMF (8 ml) hinzugegeben, und das Gemisch wurde 2 h bei 60 °C gerührt und anschließend über Nacht unter Rühren auf RT kommen gelassen. An- schließend wurde das Gemisch mit Ethylacetat und Wasser (jeweils 150 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (80 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid- Lösung (200 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge Ultra, Cyclohexan/ Ethylacetat-Gradient 97:3— > 7:3, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 1.06 g (95% Reinheit, 56% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.60 min; MS (ESIpos): m/z = 643/645 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.150 (0.03), -0.009 (0.28), 0.007 (0.25), 0.145 (0.03), 0.888 (0.03), 1.156 (0.13), 1.174 (0.26), 1.192 (0.14), 1.198 (0.07), 1.255 (0.06), 1.31 1 (0.14), 1.358 (16.00), 1.396 (2.64), 1.514 (0.07), 1.988 (0.58), 2.059 (0.18), 2.073 (0.22), 2.089 (0.23), 2.109 (0.21 ), 2.156 (1.59), 2.320 (0.10), 2.365 (0.04), 2.669 (0.04), 2.709 (0.04), 2.727 (0.17), 3.721 (0.22), 3.805 (0.22), 4.002 (0.05), 4.020 (0.12), 4.038 (0.12), 4.056 (0.05), 7.268 (0.1 1 ), 7.279 (0.13), 7.292 (0.23), 7.303 (0.24), 7.317 (0.19), 7.328 (0.16), 7.402 (0.22), 7.492 (0.28), 7.502 (0.83), 7.510 (0.79), 7.521 (2.70), 7.532 (1.1 1 ), 7.568 (0.08), 7.586 (0.06), 7.733 (0.08), 7.751 (0.08), 7.841 (0.43), 7.846 (0.38), 7.863 (0.66), 7.869 (0.62), 7.936 (1.19), 7.958 (0.76), 8.215 (0.03), 8.935 (0.21 ), 8.949 (0.39), 8.962 (0.21 ).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.95 (t, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.70 (br. s, 1 H), 7.56-7.47 (m, 5H), 7.45-7.36 (m, 1 H), 7.35-7.25 (m, 1 H), 3.88-3.66 (m, 3H), 2.21-1.91 (m, 7H), 1.36 (s, 9H).
Trennung der Enantiomere:
Die Titelverbindung (925 mg) wurde in Isopropanol (20 ml) gelöst und mittels präparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 250A und 251A) [Säule: Daicel Chiralpak AD-H, 5 μηη, 250 mm x 20 mm; Fluss: 20 ml/min; Detektion: 220 nm; Temperatur: 23 °C; Injektion: 0.2 ml; Eluent: 70% Heptan / 30% Isopropanol; isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt, und der jeweilige Rückstand wurde in Acetonitril/Wasser lyophilisiert.
Beispiel 250A
(-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlor-3,6-difluor- phenyl)pentanoat (Enantiomer 1)
Bei der in Beispiel 249A beschriebenen Enantiomerentrennung wurden 289 mg (100% Reinheit, ee-Wert 99%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
Rt = 1 .40 min (chirale analytische HPLC; Daicel Chiralpak AD-3, 3 μηη, 50 mm x 4.6 mm, Eluent Heptan / Isopropanol 80:20; Fluss 1 ml/min; Detektion 220 nM) [a]D 20 = -20.2°, 589 nm, c = 0.35 g/100 ml, Chloroform
LC-MS (Methode 1 ): Rt = 2.63 min; MS (ESIpos): m/z = 643/645 [M+H]+
1 H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1 .310 (0.15), 1 .358 (16.00), 1 .979 (0.12), 2.072 (0.24), 2.088 (0.24), 2.155 (1 .70), 3.719 (0.24), 3.805 (0.25), 7.302 (0.26), 7.317 (0.20), 7.400 (0.24), 7.502 (0.87), 7.510 (0.83), 7.521 (3.00), 7.841 (0.45), 7.846 (0.39), 7.863 (0.68), 7.868 (0.64), 7.935 (1 .27), 7.958 (0.78), 8.948 (0.41 ).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.95 (t, 1 H), 7.94 (d, 1 H), 7.86 (dd, 1 H), 7.71 (br. s, 1 H), 7.56-7.48 (m, 5H), 7.45-7.36 (m, 1 H), 7.35-7.26 (m, 1 H), 3.90-3.66 (m, 3H), 2.23-1 .89 (m, 7H), 1 .36 (s, 9H).
Beispiel 251A
(+)-ie f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlor-3,6-difluor- phenyl)pentanoat (Enantiomer 2)
Bei der in Beispiel 249A beschriebenen Enantiomerentrennung wurden 298 mg (97% Reinheit, ee-Wert 98%) der Titelverbindung als später eluierendes Enantiomer erhalten.
Rt = 1 .51 min (chirale analytische HPLC; Daicel Chiralpak AD-3, 3 μηη, 50 mm x 4.6 mm, Eluent Heptan / Isopropanol 80:20; Fluss 1 ml/min; Detektion 220 nM)
[a]D 20 = +19.1 °, 589 nm, c = 0.38 g/100 ml, Chloroform
LC-MS (Methode 1 ): Rt = 2.63 min; MS (ESIpos): m/z = 643/645 [M+H]+
1 H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.846 (0.07), 0.859 (0.09), 1 .087 (0.05), 1 .103 (0.06), 1 .199 (0.08), 1 .256 (0.09), 1 .312 (0.15), 1 .359 (16.00), 1 .515 (0.08), 1 .984 (0.14), 2.074 (0.28), 2.090 (0.28), 2.1 10 (0.26), 2.157 (2.02), 2.323 (0.07), 2.671 (0.04), 3.723 (0.27), 3.806 (0.29), 7.293 (0.28), 7.304 (0.30), 7.318 (0.22), 7.329 (0.20), 7.403 (0.27), 7.493 (0.32), 7.503 (0.92), 7.51 1 (0.88), 7.523 (3.28), 7.722 (0.07), 7.843 (0.44), 7.865 (0.68), 7.869 (0.67), 7.937 (1 .19), 7.959 (0.74), 8.951 (0.47).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.95 (t, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.72 (br. s, 1 H), 7.57-7.47 (m, 5H), 7.45-7.36 (m, 1 H), 7.34-7.24 (m, 1 H), 3.90-3.65 (m, 3H), 2.22-1 .89 (m, 7H), 1 .36 (s, 9H).
Beispiel 252A
(+/-)-fe/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchm^
pentanoat (Racemat)

Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (2.30 g, 6.73 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (25 ml) wurden HATU (3.07 g, 8.08 mmol) und DIPEA (2.8 ml, 16 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-5-amino-4-(2-fluorphenyl)pentanoat (3.00 g, 60% Reinheit, 6.73 mmol, Beispiel 129A), gelöst in DMF (15 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Anschließend wurde das Gemisch mit Ethylacetat und Wasser (jeweils 150 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (80 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung (200 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge Ultra, Cyclohexan/ Ethylacetat-Gradient 97:3— > 7:3, Isolera One) vorgereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand in Acetonitril (40 ml) gelöst und mittels präparativer HPLC [Säule: Chromatorex Spring Column C18, 5 μηη, 370 mm x 100 mm; Fluss: 250 ml/min; Detekti- on: 210 nm; Temperatur: 22 °C; Injektion: 18 ml; Acetonitril-Wasser-Gradient 1 :1— > 9:1 ; Laufzeit 45 min] nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 2.49 g (98% Reinheit, 61 % d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.33 min; MS (ESIpos): m/z = 591/593 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.022 (0.16), 0.007 (0.06), 0.851 (0.03), 1.205 (0.07), 1.230 (0.37), 1.291 (0.07), 1.321 (0.08), 1.366 (16.00), 1.522 (0.07), 1.654 (0.02), 1.679 (0.02), 1.685 (0.03), 1.691 (0.04), 1.704 (0.03), 1.71 1 (0.04), 1.741 (0.09), 1.749 (0.1 1 ), 1.757 (0.24), 1.766 (0.12), 1.774 (0.15), 1.793 (0.16), 1.810 (0.21 ), 1.818 (0.19), 1.835 (0.15), 1.869 (0.04), 1.910 (0.03), 1.925 (0.03), 1.949 (0.03), 1.957 (0.04), 1.964 (0.03), 1.999 (0.08), 2.021 (0.17),
2.037 (0.25), 2.049 (0.25), 2.074 (1.23), 2.088 (0.95), 2.1 10 (0.59), 2.297 (0.02), 2.327 (0.02), 2.365 (0.02), 2.669 (0.01 ), 2.709 (0.01 ), 3.314 (3.22), 3.339 (0.19), 3.352 (0.19), 3.361 (0.18), 3.387 (0.07), 3.584 (0.08), 3.600 (0.17), 3.616 (0.08), 3.633 (0.08), 3.647 (0.15), 3.665 (0.20), 3.680 (0.25), 3.693 (0.14), 3.724 (0.12), 3.742 (0.18), 3.761 (0.18), 3.779 (0.15), 3.797 (0.1 1 ), 3.805 (0.06), 3.817 (0.04), 5.210 (0.02), 5.308 (0.01 ), 5.364 (0.03), 5.369 (0.03), 5.379 (0.03), 5.384 (0.03), 5.840 (0.02), 5.851 (0.02), 7.142 (0.23), 7.163 (0.37), 7.188 (0.45), 7.203 (0.42), 7.222 (0.30), 7.283 (0.24), 7.298 (0.24), 7.320 (0.10), 7.340 (0.05), 7.396 (0.25), 7.399 (0.25), 7.415 (0.45), 7.433 (0.22), 7.486 (0.24), 7.498 (0.71 ), 7.505 (0.76), 7.516 (2.97), 7.527 (0.95), 7.679 (0.06), 7.829 (0.36), 7.834 (0.32), 7.851 (0.56), 7.856 (0.52), 7.926 (1.08), 7.948 (0.69), 8.850 (0.24), 8.864 (0.42), 8.878 (0.23), 1 1.682 (0.15).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.86 (t, 1 H), 7.93 (d, 1 H), 7.84 (dd, 1 H), 7.68 (br. s, 1 H), 7.55-7.47 (m, 5H), 7.44-7.38 (m, 1 H), 7.36-7.25 (m, 1 H), 7.24-7.12 (m, 2H), 3.84-3.71 (m, 1 H), 3.70-3.57 (m, 1 H), 3.41 -3.32 (m, 1 H), 2.21-1.96 (m, 6H), 1.86-1.72 (m, 1 H), 1.37 (s, 9H).
Beispiel 253A
(+/-)-ie f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]ami
phenyl]pentanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (2.52 g, 7.36 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (25 ml) wurden HATU (3.36 g, 8.83 mmol) und DIPEA (3.1 ml, 18 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ierf-Butyl-5-amino-4-[2-(difluormethoxy)phenyl]pentanoat (2.90 g, 80% Reinheit, 7.36 mmol, Beispiel 130A), gelöst in DMF (15 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Anschließend wurde das Gemisch mit Ethylacetat und Wasser (jeweils 150 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (80 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung (200 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Acetonitril (100 ml) in der Wärme gelöst und
mittels präparativer HPLC [Säule: Chromatorex Spring Column C18, 5 μηη, 370 mm x 100 mm; Fluss: 250 ml/min; Detektion: 210 nm; Temperatur: 22 °C; Injektion: 18 ml; Aceton itril-Wasser- Gradient 6:4— > 95:5; Laufzeit 37 min] gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 4.03 g (98% Reinheit, 84% d. Th.) der Ti- telverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.56 min; MS (ESIpos): m/z = 639/641 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.333 (1.41 ), 1.356 (16.00), 1.759 (0.42), 2.060 (1.16), 2.070 (0.62), 2.076 (0.47), 2.124 (0.42), 6.998 (0.43), 7.156 (0.47), 7.176 (0.61 ), 7.183 (0.92), 7.257 (0.45), 7.368 (0.42), 7.423 (0.47), 7.427 (0.46), 7.501 (0.72), 7.508 (0.71 ), 7.520 (2.29), 7.531 (0.99), 7.534 (0.88), 7.857 (0.56), 7.862 (0.54), 7.931 (1.04), 7.953 (0.66), 8.840 (0.43).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.84 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.68 (br. s, 1 H), 7.56-7.47 (m, 5H), 7.43 (dd, 1 H), 7.36-7.22 (m, 2H), 7.20-6.97 (m, 2H), 3.81-3.62 (m, 2H), 3.43- 3.34 (m, 1 H), 2.18-1.98 (m, 6H), 1.92-1.79 (m, 1 H), 1.36 (s, 9H).
Beispiel 254A
(+/-)-ie f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2,6-difluorphenyl)- pentanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (3.84 g, 1 1.2 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (40 ml) wurden HATU (5.12 g, 13.5 mmol) und DIPEA (4.7 ml, 27 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-5-amino-4-(2,6-difluorphenyl)pentanoat (4.00 g, 80% Reinheit, 1 1.2 mmol, Beispiel 131 A), gelöst in DMF (20 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Anschließend wurde das Gemisch mit Ethylacetat und Wasser (jeweils 150 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (80 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung (200 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Acetonitril (100 ml) gelöst und mittels präpa-
rativer HPLC [Säule: Chromatorex Spring Column C18, 5 μηη, 370 mm x 100 mm; Fluss: 250 ml/min; Detektion: 210 nm; Temperatur: 22 °C; Injektion: 18 ml; Acetonitril-Wasser-Gradient 6:4 — > 95:5; Laufzeit 37 min] gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 5.91 g (98% Reinheit, 85% d. Th.) der Titelverbin- dung erhalten.
LC-MS (Methode 1 ): Rt = 2.55 min; MS (ESIpos): m/z = 609/61 1 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.201 (0.08), 1.231 (0.03), 1.272 (0.06), 1.297 (0.04), 1.312 (0.10), 1.361 (16.00), 1.518 (0.07), 1.711 (0.04), 1.742 (0.16), 1.758 (0.40), 1.765 (0.20), 1.774 (0.16), 1.897 (0.21 ), 1.912 (0.26), 1.936 (0.23), 1.957 (0.1 1 ), 2.026 (0.09), 2.044 (0.23), 2.058 (0.29), 2.079 (0.50), 2.105 (0.62), 2.121 (1.21 ), 2.141 (1.42), 2.326 (0.04), 2.365 (0.02), 2.670 (0.02), 3.509 (0.23), 3.584 (0.16), 3.600 (0.36), 3.615 (0.15), 3.647 (0.03), 3.663 (0.03), 3.701 (0.08), 3.715 (0.15), 3.733 (0.32), 3.749 (0.50), 3.761 (0.38), 3.795 (0.23), 5.314 (0.02), 5.363 (0.02), 5.378 (0.02), 7.053 (0.56), 7.075 (1.05), 7.099 (0.65), 7.329 (0.22), 7.347 (0.28), 7.365 (0.19), 7.488 (0.30), 7.499 (0.85), 7.508 (0.85), 7.518 (3.58), 7.718 (0.08), 7.836 (0.43), 7.859 (0.68), 7.930 (1.14), 7.952 (0.72), 8.934 (0.31 ), 8.949 (0.60), 8.963 (0.30), 1 1.681 (0.05).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.95 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.72 (br. s, 1 H), 7.55-7.46 (m, 5H), 7.39-7.29 (m, 1 H), 7.08 (t, 2H), 3.85-3.67 (m, 2H), 3.57-3.45 (m, 1 H), 2.22- 2.01 (m, 6H), 1.98-1.85 (m, 1 H), 1.36 (s, 9H).
Beispiel 255A
(+/-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlor-3-fluor- phenyl)pentanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (1.03 g, 3.00 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (12 ml) wurden HATU (1.37 g, 3.60 mmol) und DIPEA (1.3 ml, 7.2 mmol) gegeben und das Gemisch wurde 15 min bei RT gerührt. Anschließend wurde (+/-)-ierf-Butyl-5-amino-4-(2-chlor-3-fluorphenyl)pentanoat (905 mg,
3.00 mmol, nicht reinheitskomgiert, Beispiel 132A), gelöst in DMF (9 ml) hinzugegeben, und das Gemisch wurde 3 h bei 60 °C gerührt. Anschließend wurde das Gemisch mit Ethylacetat und Wasser (jeweils 100 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (80 ml) extrahiert. Die vereinigten organischen Phasen wurden mit ge- sättigter wässriger Natriumchlorid-Lösung (100 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash- Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge Ultra, Cyclohexan/Ethylacetat- Gradient 98:2— > 8:2, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 855 mg (97% Reinheit, 44% d. Th.) der Titelver- bindung erhalten.
LC-MS (Methode 1 ): Rt = 2.59 min; MS (ESIpos): m/z = 625/627 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.151 (0.02), 0.007 (0.14), 0.144 (0.02), 0.887 (0.03), 1.156 (0.19), 1.174 (0.37), 1.192 (0.19), 1.206 (0.07), 1.232 (0.04), 1.284 (0.08), 1.308 (0.04), 1.328 (0.12), 1.366 (16.00), 1.395 (0.14), 1.522 (0.07), 1.786 (0.05), 1.805 (0.1 1 ), 1.820 (0.15), 1.837 (0.19), 1.987 (0.69), 2.018 (0.08), 2.039 (0.17), 2.052 (0.21 ), 2.068 (0.21 ), 2.095 (1.18), 2.1 10 (0.92), 2.131 (0.68), 2.143 (0.72), 2.265 (0.04), 2.320 (0.06), 2.365 (0.02), 2.669 (0.02), 2.709 (0.02), 3.630 (0.22), 3.661 (0.10), 3.743 (0.27), 4.002 (0.05), 4.020 (0.16), 4.038 (0.16), 4.055 (0.05), 7.142 (0.03), 7.279 (0.16), 7.299 (0.32), 7.320 (0.21 ), 7.362 (0.26), 7.380 (0.55), 7.396 (0.23), 7.415 (0.26), 7.430 (0.23), 7.449 (0.08), 7.476 (0.10), 7.490 (0.27), 7.499 (0.80), 7.508 (0.75), 7.519 (2.46), 7.530 (1.09), 7.646 (0.06), 7.830 (0.39), 7.836 (0.35), 7.853 (0.59), 7.858 (0.56), 7.929 (1.1 1 ), 7.951 (0.72), 8.849 (0.24), 8.863 (0.47), 8.878 (0.23).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.87 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.65 (br. s, 1 H), 7.56-7.47 (m, 5H), 7.46-7.35 (m, 2H), 7.34-7.26 (m, 1 H), 3.74 (br. s, 2H), 3.67-3.58 (m, 1 H), 2.21-2.00 (m, 6H), 1.89-1.77 (m, 1 H), 1.37 (s, 9H).
Trennung der Enantiomere:
Die Titelverbindung (715 mg) wurde in Methanol (30 ml) gelöst und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 256A und 257A) [Säule: Daicel Chi- ralcel OZ-H, 5 μηη, 250 mm x 30 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 0.5 ml; Eluent: 85% Kohlendioxid / 15% Methanol; Laufzeit 12 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet.
Beispiel 256A
(-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlor-3-fluor- phenyl)pentanoat (Enantiomer 1)
Bei der in Beispiel 255A beschriebenen Enantiomerentrennung wurden 298 mg (100% Reinheit, ee-Wert 96%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -5.1 °, 436 nm, c = 0.46 g/100 ml, Chloroform
LC-MS (Methode 1 ): Rt = 2.60 min; MS (ESIpos): m/z = 625/627 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.208 (0.07), 1.286 (0.07), 1.329 (0.13), 1.368 (16.00), 1.524 (0.07), 1.821 (0.18), 1.839 (0.23), 2.040 (0.19), 2.054 (0.23), 2.074 (0.29), 2.097 (1.32), 2.1 1 1 (1.07), 2.133 (0.84), 2.145 (0.88), 2.266 (0.04), 2.329 (0.03), 2.670 (0.03), 3.632 (0.26), 3.746 (0.33), 7.281 (0.19), 7.301 (0.38), 7.323 (0.26), 7.364 (0.31 ), 7.382 (0.64), 7.398 (0.28), 7.416 (0.32), 7.431 (0.27), 7.491 (0.32), 7.502 (0.89), 7.509 (0.88), 7.521 (2.85), 7.650 (0.07), 7.833 (0.40), 7.838 (0.39), 7.855 (0.62), 7.860 (0.61 ), 7.931 (1.12), 7.953 (0.72), 8.852 (0.28), 8.866 (0.53), 8.880 (0.27).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.87 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.65 (br. s, 1 H), 7.56-7.47 (m, 5H), 7.46-7.35 (m, 2H), 7.34-7.26 (m, 1 H), 3.75 (br. s, 2H), 3.67-3.58 (m, 1 H), 2.22-2.00 (m, 6H), 1.89-1.76 (m, 1 H), 1.37 (s, 9H).
Beispiel 257A
(+)-fe/f-Butyl-5-{[(6-brom-3-methyl-2-p^
phenyl)pentanoat
Bei der in Beispiel 255A beschriebenen Enantiomerentrennung wurden 310 mg (100% Reinheit, ee-Wert 96%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = +8.4°, 436 nm, c = 0.47 g/100 ml, Chloroform
LC-MS (Methode 1 ): Rt = 2.60 min; MS (ESIpos): m/z = 625/627 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.208 (0.07), 1.285 (0.08), 1.329 (0.15), 1.367 (16.00), 1.524 (0.07), 1.837 (0.24), 2.053 (0.25), 2.074 (0.30), 2.096 (1.35), 2.1 10 (1.1 1 ), 2.132 (0.89), 2.143 (0.92), 2.328 (0.04), 2.367 (0.04), 2.671 (0.04), 3.630 (0.28), 3.743 (0.35), 7.281 (0.20), 7.301 (0.39), 7.322 (0.27), 7.363 (0.33), 7.381 (0.66), 7.398 (0.29), 7.416 (0.33), 7.431 (0.28), 7.502 (0.93), 7.509 (0.95), 7.521 (3.01 ), 7.651 (0.08), 7.833 (0.42), 7.837 (0.39), 7.855 (0.64), 7.860 (0.61 ), 7.930 (1.10), 7.953 (0.70), 8.851 (0.29), 8.865 (0.54), 8.879 (0.27).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.86 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.65 (br. s, 1 H), 7.56-7.47 (m, 5H), 7.46-7.35 (m, 2H), 7.34-7.26 (m, 1 H), 3.74 (br. s, 2H), 3.68-3.55 (m, 1 H), 2.19-2.00 (m, 6H), 1.89-1.76 (m, 1 H), 1.37 (s, 9H).
Beispiel 258A
(+/-)-ie f-Butyl-/V-[2-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbon
ethyl]-/V-methylglycinat (Racemat)

Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (428 mg, 1.25 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (5 ml) wurden HATU (713 mg, 1.87 mmol) und DIPEA (650 μΙ, 3.7 mmol) gegeben, und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ierf-Butyl-/V-[2-amino-1-(2-chlorphenyl)ethyl]-/V-methylglycinat (800 mg, 41 % Reinheit, 1.01 mmol, Beispiel 133A), gelöst in DMF (3 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Anschließend wurde das Gemisch mit Ethylacetat und Wasser (jeweils 20 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase zweimal mit Ethylacetat (20 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage, Cyclohexan/Ethylacetat 85:15, Isolera One) vorgereinigt. Anschließend wurde mittels präparativer HPLC (Methode 16) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert. Es wurden 150 mg (98% Reinheit, 19% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.68 min; MS (ESIpos): m/z = 622/624 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.85 (br. s, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.72 (br. d, 2H), 7.58-7.37 (m, 9H), 4.99 (br. s, 1 H), 4.14 (br. s, 1 H), 4.00 (br. s, 1 H), 3.72 (br. s, 1 H), 3.53 (br. s, 1 H), 2.59 (br. s, 3H), 2.10 (s, 3H), 1.43 (s, 9H).
Beispiel 259A
(+/-)-ie f-Butyl-/V-[2-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbon
ethyl]glycinat (Racemat)

Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (428 mg, 1.25 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (8 ml) wurden HATU (1.10 g, 2.89 mmol) und DIPEA (1.0 ml, 5.8 mmol) gegeben, und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-/V-[2-amino-1-(2-chlorphenyl)ethyl]glycinat (1.64 g, 50% Reinheit, 2.89 mmol, Beispiel 134A), gelöst in DMF (4 ml) hinzugegeben, und das Ge- misch wurde über Nacht bei 60 °C gerührt. Anschließend wurde das Gemisch mit Ethylacetat und Wasser (jeweils 20 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase zweimal mit Ethylacetat (20 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage, Cyclohexan/Ethylacetat 85:15, Isolera One) vorgereinigt. Anschließend wurde mittels präparativer HPLC (Methode 16) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert. Es wurden 300 mg (98% Reinheit, 25% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.45 min; MS (ESIpos): m/z = 608/610 [M+H]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.99 (t, 1 H), 8.03-7.80 (m, 3H), 7.77-7.42 (m, 9H), 4.99 (br. s, 1 H), 4.18-3.99 (br. m, 2H), 3.78-3.64 (br. s, 2H), 2.09 (br. s, 3H), 1.44 (s, 9H).
Beispiel 260A
(+/-)-Ethyl-[2-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]am
acetat (Racemat)

Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (1 .58 g, 4.63 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (15 ml) wurden HATU (2.1 1 g, 5.55 mmol) und DI PEA (2.7 ml, 16 mmol) gegeben, und das Gemisch wurde 15 min bei RT gerührt. Anschließend wurde (+/-)-Ethyl-[2-amino-1 -(2-chlorphenyl)ethoxy]acetat-Hydrochlorid (1 .89 g, 72% Reinheit, 4.63 mmol, Beispiel 137A), gelöst in DMF (10 ml) hinzugegeben, und das Ge- misch wurde 1 h bei 60 °C gerührt und danach 16 h bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mit Ethylacetat und Wasser (jeweils 80 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase zweimal mit Ethylacetat (80 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung (100 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge Ultra, Cyclohexan/ Ethylacetat-Gradient 93:7— > 7:3, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 1 .1 1 g (92% Reinheit, 38% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.41 min; MS (ESIpos): m/z = 581/583 [M+H]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.97 (t, 1 H), 7.96 (d, 1 H), 7.90-7.84 (m, 2H), 7.62-7.47 (m, 7H), 7.47-7.36 (m, 2H), 5.20 (t, 1 H), 4.21 -3.99 (m, 4H), 3.78 (br. s, 2H), 2.26 (s, 3H), 1.13 (t, 3H).
Trennung der Enantiomere:
Die Titelverbindung (1 .10 g) wurde in einem Ethanol-Acetonitril-Gemisch (60 ml) gelöst und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 261A und 262A) [Säule: Daicel Chiralpak AD-H, 5 [Jim , 250 mm x 30 mm; Fluss: 125 ml/min; Detektion: 210 nm; Temperatur: 38 °C; Injektion: 1 .6 ml; Eluent: 67% Kohlendioxid / 33% Ethanol -> 50% Koh-
lendioxid / 50% Ethanol; Laufzeit 12 min]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet.
Beispiel 261A
(+)-Ethyl-[2-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-1-(2-chlorphenyl)ethoxy]- acetat (Enantiomer 1)
Bei der in Beispiel 260A beschriebenen Enantiomerentrennung wurden 440 mg (94% Reinheit, ee-Wert 100%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = +24.9°, 489 nm, c = 1.00 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.25 min; MS (ESIpos): m/z = 581/583 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.1 17 (6.52), 1.135 (13.59), 1.152 (6.76), 2.239 (0.40), 2.261 (16.00), 2.321 (0.41 ), 3.782 (1.32), 4.001 (2.96), 4.042 (5.07), 4.050 (2.37), 4.068 (6.37), 4.085 (6.28), 4.103 (2.07), 4.137 (4.91 ), 4.178 (2.81 ), 5.188 (1.29), 5.203 (2.36), 5.217 (1.26), 7.372 (0.70), 7.376 (0.76), 7.390 (2.05), 7.394 (2.03), 7.409 (2.14), 7.414 (2.18), 7.419 (1.71 ), 7.422 (1.81 ), 7.438 (2.33), 7.440 (2.37), 7.456 (1.05), 7.491 (3.36), 7.495 (3.57), 7.513 (7.07), 7.533 (5.81 ), 7.556 (6.72), 7.575 (2.46), 7.583 (3.18), 7.588 (2.62), 7.603 (2.30), 7.607 (2.01 ), 7.854 (2.78), 7.873 (4.22), 7.948 (4.77), 7.971 (2.98), 8.951 (1.24), 8.965 (2.55), 8.979 (1.23).
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.97 (t, 1 H), 7.96 (d, 1 H), 7.89-7.83 (m, 2H), 7.64-7.47 (m, 7H), 7.47-7.35 (m, 2H), 5.20 (t, 1 H), 4.19-3.98 (m, 4H), 3.78 (br. s, 2H), 2.26 (s, 3H), 1.13 (t, 3H).
Beispiel 262A
(-)-Ethyl-[2-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-1-(2-chlorphenyl)ethoxy]- acetat (Enantiomer 2)
Bei der in Beispiel 260A beschriebenen Enantiomerentrennung wurden 415 mg (100% Reinheit, ee-Wert 100%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = -28.3°, 489 nm, c = 1.00 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.25 min; MS (ESIpos): m/z = 581/583 [M-H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (0.63), 1.056 (0.45), 1.1 17 (7.22), 1.135 (15.06), 1.153 (7.43), 1.235 (0.42), 2.261 (16.00), 2.524 (0.66), 3.781 (1.35), 4.001 (2.99), 4.041 (5.13), 4.050 (2.50), 4.068 (6.74), 4.086 (6.66), 4.103 (2.18), 4.137 (5.01 ), 4.177 (2.86), 5.188 (1 .31 ), 5.202 (2.36), 5.216 (1.26), 7.372 (0.74), 7.376 (0.87), 7.391 (2.15), 7.395 (2.25), 7.409 (2.27), 7.414 (2.40), 7.419 (1.79), 7.423 (1.97), 7.438 (2.31 ), 7.441 (2.57), 7.457 (1.07), 7.491 (3.48), 7.495 (3.85), 7.514 (7.54), 7.534 (5.99), 7.555 (6.98), 7.559 (5.88), 7.575 (2.51 ), 7.583 (3.31 ), 7.588 (2.71 ), 7.602 (2.27), 7.606 (2.08), 7.855 (3.17), 7.873 (4.31 ), 7.948 (5.06), 7.971 (3.14), 8.951 (1.27), 8.965 (2.55), 8.980 (1.20).
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.97 (t, 1 H), 7.96 (d, 1 H), 7.89-7.83 (m, 2H), 7.62-7.47 (m, 7H), 7.47-7.36 (m, 2H), 5.20 (t, 1 H), 4.19-3.98 (m, 4H), 3.78 (br. s, 2H), 2.26 (s, 3H), 1.13 (t, 3H).
Beispiel 263A
(+/-)-Methyl-{[2-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]am
sulfanyl}acetat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (1.44 g, 4.20 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (8 ml) wurden HATU (2.39 g, 6.29 mmol) und DIPEA (2.2 ml, 13 mmol) gegeben, und das Gemisch wurde 30 min bei RT ge- rührt. Anschließend wurde (+/-)-Methyl-{[2-amino-1-(2-chlorphenyl)ethyl]sulfanyl}acetat (3.27 g, 50% Reinheit, 6.29 mmol), Beispiel 138A), gelöst in DMF (4 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethylacetat und Wasser (jeweils 20 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase zweimal mit Ethylacetat (20 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Acetonitril gelöst und mittels präparativer HPLC (Methode 16) vorgereinigt. Anschließend wurde mittels Flash-Säulenchromatographie (340 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat-Gradient 93:7— > 6:4, Isolera One) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 388 mg (87% Reinheit, 14% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.20 min; MS (ESIpos): m/z = 583/585 [M+H]+
Beispiel 264A
(+/-)-Methyl-{[2-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbon
sulfonyl}acetat (Racemat)

Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (63 mg, 183 μηηοΙ, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (1.5 ml) wurden HATU (83 mg, 219 μηηοΙ) und DIPEA (76 μΙ, 440 μηηοΙ) gegeben, und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-Methyl-{[2-amino-1-(2-chlorphenyl)ethyl]sulfonyl}acetat-Hydrochlorid (60 mg, 183 μηηοΙ, Beispiel 141A), gelöst in DMF (0.5 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethylacetat und Wasser (jeweils 150 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (80 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung (200 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels präparativer HPLC (Methode 17) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 66 mg (93% Reinheit, 55% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.04 min; MS (ESIpos): m/z = 615/617 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 9.14 (t, 1 H), 7.93 (d, 1 H), 7.84 (dd, 1 H), 7.75-7.56 (m, 3H), 7.55-7.41 (m, 7H), 5.75 (dd, 1 H), 4.81 (d, 1 H), 4.49 (d, 1 H), 4.41 (td, 1 H), 4.26-4.10 (m, 1 H), 3.75 (s, 3H), 2.02 (br. s, 3H).
Beispiel 265A
(+/-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(6-chlor-2,3-difluor- phenyl)pentanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (621 mg, 1.82 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (8 ml) wurden HATU (829 mg, 2.18 mmol) und DIPEA (760 μΙ, 4.4 mmol) gegeben und das Gemisch wurde 15 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-5-amino-4-(6-chlor-2,3- difluorphenyl)pentanoat (600 mg, 97% Reinheit, 1.82 mmol, Beispiel 142A), gelöst in DMF (4 ml) hinzugegeben, und das Gemisch wurde 3 h bei 60 °C, gefolgt von 2 h bei RT gerührt. Anschließend wurde das Gemisch mit Ethylacetat und Wasser (jeweils 100 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (80 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung (100 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge Ultra, Cyclohexan/ Ethylacetat-Gradient 97:3— > 7:3, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 154 mg (100% Reinheit, 13% d. Th.) einer ersten Charge Titelverbindung erhalten. Außerdem wurde eine vorgereinigte Fraktion erhalten, welche in Acetonitril aufgenommen und mittels präparativer HPLC (Methode 15) nachgereinigt wurde. Daraus wurden 497 mg (100% Reinheit, 42% d. Th., siehe Analytik) einer zweiten Charge Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.37 min; MS (ESIpos): m/z = 643/645 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.95 (t, 1 H), 7.94 (d, 1 H), 7.86 (dd, 1 H), 7.69 (br. s, 1 H), 7.59-7.47 (m, 5H), 7.46-7.32 (m, 2H), 3.81 (br. s, 2H), 3.72 (br. s, 1 H), 2.22-1.93 (m, 7H), 1.36 (s, 9H).
Trennung der Enantiomere:
Die Titelverbindung (480 mg) wurde in Methanol (25 ml) gelöst und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 266A und 267A) [Säule: Daicel Chi- ralpak AD SFC, 5 μηη, 250 mm x 20 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur:
40 °C; Injektion: 0.2 ml; Eluent: 75% Kohlendioxid / 25% Methanol; Laufzeit 4 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert.
Beispiel 266A
(-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(6-chlor-2,3-difluor- phenyl)pentanoat (Enantiomer 1)
Bei der in Beispiel 265A beschriebenen Enantiomerentrennung wurden 154 mg (100% Reinheit, ee-Wert 100%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -32.8°, 589 nm, c = 0.29 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.61 min; MS (ESIpos): m/z = 643/645 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.95 (t, 1 H), 7.94 (d, 1 H), 7.86 (dd, 1 H), 7.72 (br. s, 1 H), 7.57-7.46 (m, 5H), 7.46-7.32 (m, 2H), 3.91-3.63 (m, 3H), 2.25-1.86 (m, 7H), 1.36 (s, 9H).
Beispiel 267A
(+)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(6-chlor-2,3-difluor- phenyl)pentanoat (Enantiomer 2)
Bei der in Beispiel 265A beschriebenen Enantiomerentrennung wurden 162 mg (97% Reinheit, ee-Wert 93%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = +28.7°, 589 nm, c = 0.39 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.60 min; MS (ESIpos): m/z = 643/645 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.95 (t, 1 H), 7.94 (d, 1 H), 7.86 (dd, 1 H), 7.72 (br. s, 1 H), 7.55-7.46 (m, 5H), 7.46-7.33 (m, 2H), 3.89-3.65 (m, 3H), 2.25-1.86 (m, 7H), 1.36 (s, 9H).
Beispiel 268A
(+/-)-Methyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-[2-(trifluormethyl)- phenyl]pentanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (5.26 g, 15.4 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (60 ml) wurden HATU (7.02 g, 18.5 mmol) und DIPEA (6.4 ml, 37 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-Methyl-5-amino-4-[2-(trifluormethyl)phenyl]pentanoat (5.50 g, 77% Reinheit, 15.4 mmol, Beispiel 143A), gelöst in DMF (30 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethylacetat und Wasser (jeweils 150 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (80 ml) extrahiert. Die vereinigten organischen Pha- sen wurden mit gesättigter wässriger Natriumchlorid-Lösung (200 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (340 g Kieselgel Biotage Snap-Cartridge KP- Sil, Cyclohexan/ Ethylacetat-Gradient 97:3— > 7:3, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 4.31 g (96% Reinheit, 45% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.33 min; MS (ESIpos): m/z = 599/601 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.92 (t, 1 H), 7.95 (d, 1 H), 7.85 (dd, 1 H), 7.80-7.60 (m, 4H), 7.58-7.45 (m, 6H), 3.85-3.72 (m, 1 H), 3.72-3.62 (m, 1 H), 3.52 (s, 3H), 3.40-3.30 (m, 1 H), 2.26-2.04 (m, 6H), 2.02-1.89 (m, 1 H). Beispiel 269A
(+/-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(5-fluor-2-methyl- phenyl)pentanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (675 mg, 1.97 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (9 ml) wurden HATU (899 mg, 2.37 mmol) und DIPEA (820 μΙ, 4.7 mmol) gegeben, und das Gemisch wurde 15 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-5-amino-4-(5-fluor-2-methylphenyl)pentanoat (600 mg, 92% Reinheit, 1.97 mmol, Beispiel 144A), gelöst in DMF (4.8 ml) hinzugegeben, und das Gemisch wurde 3 h bei 60 °C, gefolgt von zwei Tagen bei RT gerührt. Anschließend wurde das Gemisch mit Ethylacetat und Wasser (jeweils 100 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (80 ml) extrahiert. Die vereinigten or- ganischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung (100 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap- Cartridge Ultra, Cyclohexan/ Ethylacetat-Gradient 97:3— > 7:3, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 625 mg (100% Reinheit, 52% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.61 min; MS (ESIpos): m/z = 605/607 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.889 (0.03), 1.156 (0.17), 1.174 (0.34), 1.192 (0.17), 1.207 (0.07), 1.233 (0.06), 1.303 (0.07), 1.336 (0.1 1 ), 1.367 (16.00), 1.396 (0.60), 1.524 (0.07), 1.744 (0.05), 1.761 (0.12), 1.778 (0.15), 1.786 (0.15), 1.795 (0.19), 1.819 (0.16), 1.834 (0.08), 1.988 (0.79), 2.001 (0.21 ), 2.018 (0.22), 2.038 (0.21 ), 2.051 (0.22), 2.069 (0.82), 2.086 (1.18), 2.104 (0.81 ), 2.292 (2.47), 2.669 (0.03), 2.730 (0.08), 2.889 (0.07), 3.510 (0.1 1 ), 3.523 (0.17), 3.543 (0.19), 3.557 (0.22), 3.570 (0.13), 3.708 (0.15), 3.725 (0.22), 3.745 (0.19), 3.759 (0.16), 3.779 (0.10), 4.002 (0.05), 4.020 (0.15), 4.038 (0.15), 4.056 (0.05), 6.918 (0.14), 6.938 (0.27), 6.954 (0.15), 7.155 (0.34), 7.162 (0.35), 7.189 (0.63), 7.204 (0.37), 7.225 (0.27), 7.489 (0.25), 7.499 (0.76), 7.507 (0.72), 7.518 (2.36), 7.529 (1.05), 7.647 (0.10), 7.834 (0.40), 7.839 (0.37), 7.856 (0.60), 7.861 (0.59), 7.932 (1.08), 7.955 (0.69), 8.829 (0.23), 8.843 (0.35), 8.858 (0.22).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.84 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.65 (br. s, 1 H), 7.56-7.45 (m, 5H), 7.25-7.13 (m, 2H), 6.94 (td, 1 H), 3.81-3.68 (m, 1 H), 3.59-3.47 (m, 1 H), 3.38-
3.29 (m, 1 H, teilweise verdeckt), 2.29 (s, 3H), 2.18-1.94 (m, 6H), 1.86-1.72 (m, 1 H), 1.37 (s, 9H).
Trennung der Enantiomere:
Die Titelverbindung (400 mg) wurde in Methanol (18 ml) gelöst und mittels praparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 270A und 271A) [Säule: Daicel Chi- ralcel OX-H, 5 μηη, 250 mm x 20 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 0.3 ml; Eluent: 80% Kohlendioxid / 20% Ethanol; Laufzeit 6 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert.
Beispiel 270A
(-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(5-fluor-2-methyl- phenyl)pentanoat (Enantiomer 1)
Bei der in Beispiel 269A beschriebenen Enantiomerentrennung wurden 152 mg (100% Reinheit, ee-Wert 100%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -13.7°, 589 nm, c = 0.38 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.73 min; MS (ESIpos): m/z = 605/607 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.368 (16.00), 2.070 (0.80), 2.087 (1.16), 2.105 (0.78), 2.293 (2.42), 7.190 (0.62), 7.500 (0.72), 7.508 (0.68), 7.519 (2.28), 7.530 (1.02), 7.534 (0.89), 7.857 (0.58), 7.862 (0.57), 7.933 (1.05), 7.955 (0.68).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.84 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.65 (br. s, 1 H), 7.56-7.46 (m, 5H), 7.25-7.14 (m, 2H), 6.94 (td, 1 H), 3.81-3.68 (m, 1 H), 3.59-3.49 (m, 1 H), 3.37-
3.30 (1 H, verdeckt), 2.29 (s, 3H), 2.18-1.95 (m, 6H), 1.85-1.73 (m, 1 H), 1.37 (s, 9H).
Beispiel 271A
(+)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(5-fluor-2-methyl- phenyl)pentanoat (Enantiomer 2)
Bei der in Beispiel 269A beschriebenen Enantiomerentrennung wurden 150 mg (100% Reinheit, ee-Wert 97%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = +16.9°, 589 nm, c = 0.37 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.73 min; MS (ESIpos): m/z = 605/507 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.84 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.65 (br. s, 1 H), 7.56-7.45 (m, 5H), 7.25-7.13 (m, 2H), 6.94 (td, 1 H), 3.81-3.68 (m, 1 H), 3.59-3.48 (m, 1 H), 3.37- 3.30 (1 H, verdeckt), 2.29 (s, 3H), 2.17-1.95 (m, 6H), 1.85-1.72 (m, 1 H), 1.37 (s, 9H).
Beispiel 272A
(+/-)-fe/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin^
phenyl]pentanoat (Racemat)

Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (2.56 g, 7.47 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (30 ml) wurden HATU (3.41 g, 8.96 mmol) und DIPEA (3.1 ml, 18 mmol) gegeben, und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-5-amino-4-[2-(trifluormethoxy)phenyl]pentanoat (3.00 g, 83% Reinheit, 7.47 mmol, Beispiel 145A), gelöst in DMF (10 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Anschließend wurde das Gemisch mit Ethylacetat und Wasser (jeweils 150 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (80 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung (200 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/ Ethylacetat-Gradient 97:3— > 7:3, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 3.79 g (98% Reinheit, 76 % d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.40 min; MS (ESIpos): m/z = 657/659 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.357 (16.00), 1.371 (0.55), 1.397 (3.24), 2.060 (0.44), 2.075 (1.67), 2.148 (0.51 ), 7.359 (0.52), 7.398 (0.71 ), 7.408 (0.54), 7.417 (0.67), 7.504 (0.92), 7.51 1 (0.82), 7.523 (2.03), 7.530 (1.95), 7.562 (0.55), 7.838 (0.42), 7.861 (0.65), 7.865 (0.54), 7.937 (1.07), 7.959 (0.70), 8.875 (0.47).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.87 (t, 1 H), 7.95 (d, 1 H), 7.85 (dd, 1 H), 7.67 (br s, 1 H), 7.60-7.47 (m, 6H), 7.46-7.31 (m, 3H), 3.81-3.58 (m, 2H), 3.46-3.34 (m, 1 H), 2.21-2.02 (m, 6H), 1.89-1.76 (m, 1 H), 1.36 (s, 9H).
Trennunq der Enantiomere:
Die Titelverbindung (3.50 g) wurde in Isopropanol (55 ml) in der Wärme gelöst, filtriert und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 273A und 274A) [Säule: Daicel Chiralpak OD, 20 [Jim , 360 mm x 50 mm; Fluss: 300 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 1 .0 ml; Eluent: 80% Kohlendioxid / 20% Isopropanol; Laufzeit 7.8 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet.
Beispiel 273A
(+)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-[2-(trifluormethoxy)- phenyljpentanoat (Enantiomer 1)
Bei der in Beispiel 272A beschriebenen Enantiomerentrennung wurden 1.10 g (98% Reinheit, ee-Wert 100%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = +17.0°, 589 nm, c = 0.50 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.67 min; MS (ESIpos): m/z = 657/659 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.88 (t, 1 H), 7.95 (d, 1 H), 7.85 (dd, 1 H), 7.68 (br. s, 1 H), 7.60-7.47 (m, 6H), 7.45-7.33 (m, 3H), 3.83-3.59 (m, 2H), 3.46-3.36 (m, 1 H), 2.24-2.00 (m, 6H), 1 .91 -1 .76 (m, 1 H), 1 .36 (s, 9H).
Beispiel 274A
(-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-[2-(trifluormethoxy)- phenyljpentanoat (Enantiomer 2)
Bei der in Beispiel 272A beschriebenen Enantiomerentrennung wurden 1.10 g (95% Reinheit, ee-Wert 98%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = -19.7°, 589 nm, c = 0.31 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.67 min; MS (ESIpos): m/z = 657/659 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.88 (t, 1 H), 7.95 (d, 1 H), 7.85 (dd, 1 H), 7.67 (br. s, 1 H), 7.60-7.47 (m, 6H), 7.45-7.32 (m, 3H), 3.83-3.60 (m, 2H), 3.46-3.35 (m, 1 H), 2.26-1 .99 (m, 6H), 1 .90-1 .76 (m, 1 H), 1 .36 (s, 9H).
Beispiel 275A
(+/-)-ie f-Butyl-5-{[(6-brom-3-chlor-2-phenylchinolin-4-yl)carbonyl]am
pentanoat (Racemat)
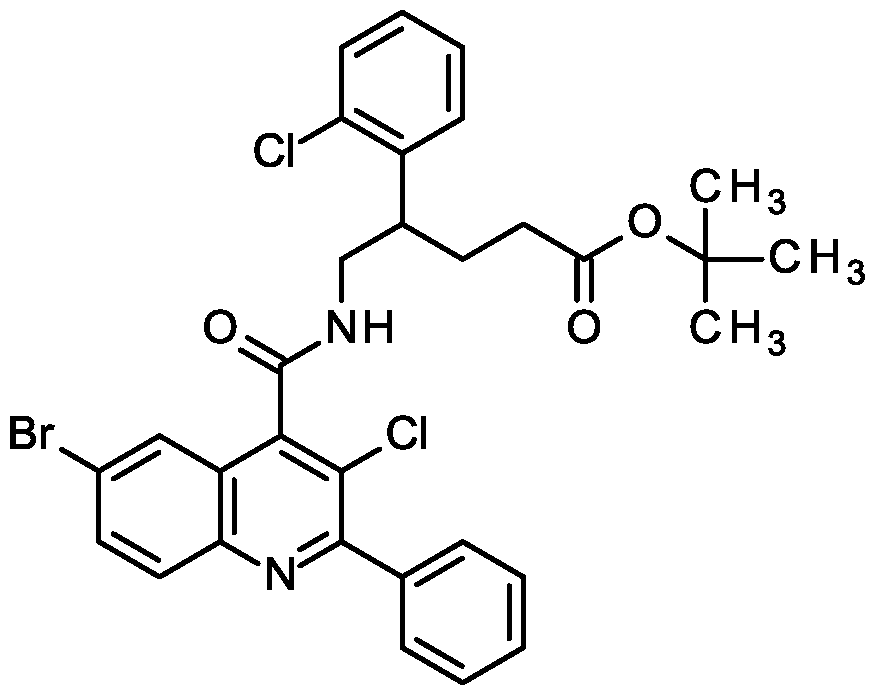
Zu einem Gemisch aus 6-Brom-3-chlor-2-phenylchinolin-4-carbonsäure (1.99 g, 5.50 mmol, nicht reinheitskorrigiert, darstellbar gemäß WO 2016 146602 A1 , S. 70, Beispiel 37A) in DMF (30 ml) wurden HATU (3.14 g, 8.25 mmol) und DIPEA (2.9 ml, 16 mmol) gegeben, und das Gemisch wurde 20 min bei RT gerührt. Anschließend wurde (+/-)-ierf-Butyl-5-amino-4-(2-chlorphenyl)pentanoat (2.34 g, 8.25 mmol, Beispiel 121A), gelöst in DMF (10 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethylacetat und Wasser (jeweils 75 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/ Ethylacetat-Gradient 97:3— > 85:15, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 2.17 g (98% Reinheit, 62 % d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.68 min; MS (ESIpos): m/z = 627/629/631 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 9.00 (t, 1 H), 8.01 (d, 1 H), 7.96 (dd, 1 H), 7.71-7.64 (m, 3H), 7.58-7.43 (m, 5H), 7.41-7.34 (m, 1 H), 7.31 -7.24 (m, 1 H), 3.77-3.67 (m, 2H), 3.66-3.56 (m, 1 H), 2.17-1.99 (m, 3H), 1.90-1.76 (m, 1 H), 1.36 (s, 9H).
Trennung der Enantiomere:
Die Titelverbindung (2.0 g) wurde in Ethanol (15 ml) gelöst und mittels präparativer HPLC an chi- raler Phase in die Enantiomere getrennt (siehe Beispiele 276A und 277A) [Säule: Daicel Chiral- pak IA, 5 μηη, 250 mm x 30 mm; Fluss: 60 ml/min; Detektion: 240 nm; Temperatur: 30 °C; Injek-
tion: 0.1 ml; Eluent: 80% Heptan / 20% Isopropanol; Laufzeit 7.5 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet.
Beispiel 276A
(-)-ie f-Butyl-5-{[(6-brom-3-chlor-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlorpheny^ pentanoat (Enantiomer 1)
Bei der in Beispiel 275A beschriebenen Enantiomerentrennung wurden 734 mg (100% Reinheit, ee-Wert 99%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
Rt = 1 .45 min (chirale analytische HPLC; Daicel Chiralpak IA-3, 3 μηη, 50 mm x 4.6 mm, Eluent Heptan / Isopropanol 80:20; Fluss 1 ml/min; Detektion 220 nM) [a]D 20 = -6.5°, 589 nm, c = 0.38 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.66 min; MS (ESIpos): m/z = 627/629/631 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 9.00 (t, 1 H), 8.01 (d, 1 H), 7.96 (dd, 1 H), 7.71 -7.63 (m, 3H), 7.58-7.42 (m, 5H), 7.38 (t, 1 H), 7.31 -7.22 (m, 1 H), 3.79-3.67 (m, 2H), 3.65-3.54 (m, 1 H), 2.15-2.00 (m, 3H), 1 .92-1.75 (m, 1 H), 1 .36 (s, 9H).
Beispiel 277A
(+)-ie/f-Butyl-5-{[(6-brom-3-chlor-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlorphenyl)- pentanoat (Enantiomer 2)
Bei der in Beispiel 275A beschriebenen Enantiomerentrennung wurden 751 mg (100% Reinheit, ee-Wert 99%) der Titelverbindung als später eluierendes Enantiomer erhalten.
Rt = 1 .92 min (chirale analytische HPLC; Daicel Chiralpak IA-3, 3 μηη, 50 mm x 4.6 mm, Eluent Heptan / Isopropanol 80:20; Fluss 1 ml/min; Detektion 220 nM)
[a]D 20 = +7.4°, 589 nm, c = 0.38 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.67 min; MS (ESIpos): m/z = 627/629/631 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 9.00 (t, 1 H), 8.01 (d, 1 H), 7.96 (dd, 1 H), 7.71 -7.63 (m, 3H), 7.58-7.43 (m, 5H), 7.38 (t, 1 H), 7.31 -7.23 (m, 1 H), 3.78-3.66 (m, 2H), 3.65-3.56 (m, 1 H), 2.17-2.00 (m, 3H), 1 .91 -1.75 (m, 1 H), 1 .36 (s, 9H).
Beispiel 278A
(+/-)-ie/f-Butyl-4-cyan-4-[2-(trifluormethyl)phenyl]butanoat (Racemat)
Zu einer Lösung aus [2-(Trifluormethyl)phenyl]acetonitril (14.8 g, 79.8 mmol, CAS-RN 3038-47-9, kommerziell verfügbar) in THF (100 ml) unter Argon wurde langsam unter Rühren eine 2 M Lösung aus LDA in THF (48 ml, 96 mmol) gegeben, wobei die Innentemperatur zwischen -70 °C und -60 °C gehalten wurde. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -70 °C abgekühlt. Anschließend wurde eine Lösung aus ief-Butyl-3-brompropanoat (15 ml, 96 mmol) in THF (70 ml) langsam unter Rühren bei -70 °C hinzu getropft. Das Gemisch wurde über Nacht weiter gerührt, wobei das Kältebad (Trockeneis/Aceton) langsam auf RT kommen gelassen wurde. Anschließend wurde das Gemisch bei ca. 0 °C langsam mit Wasser (200 ml) und Ethyl- acetat (250 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (150 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung (250 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash- Säulenchromatographie (400 g Kieselgel, Cyclohexan/Ethylacetat 9:1 ) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 18.7 g (100% Reinheit, 75% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.26 min; MS (ESIpos): m/z = 314 [M+H]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 7.91-7.75 (m, 3H), 7.62 (t, 1 H), 4.35 (dd, 1 H), 2.44-2.33 (m, 2H), 2.31-2.19 (m, 1 H), 2.18-2.06 (m, 1 H), 1.39 (s, 9H).
Beispiel 279A
(+/-)-ie/f-Butyl-5-amino-4-[2-(trifluormethyl)phenyl]pentanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-4-cyan-4-[2-(trifluormethyl)phenyl]butanoat (18.6 g, 59.5 mmol, Beispiel 278A) in ie/f-Butanol (200 ml) wurde mit Raney-Nickel (3.49 g, 59.5 mmol) versetzt und
24 h bei Normaldruck hydriert. Danach wurde der Katalysator über Kieselgur abfiltriert und zweimal mit ie f-Butanol (jeweils 50 ml) nachgewaschen. Das Filtrat wurde eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 17.8 g (82% Reinheit, 77 % d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.23 min; MS (ESIpos): m/z = 318 [M+H]+ Beispiel 280A
(+/-)-ie/f-Butyl-5-{[(6-brom-3-chlor-2-phenylchinolin-4-yl)carbonyl]amino}-4-[2-(trifluormethyl)- phenyl]pentanoat (Racemat)

Zu einem Gemisch aus 6-Brom-3-chlor-2-phenylchinolin-4-carbonsäure (500 mg, 1.38 mmol, nicht reinheitskorrigiert, darstellbar gemäß WO 2016 146602 A1 , S. 70, Beispiel 37A) in DMF (6.0 ml) wurden HATU (629 mg, 1.65 mmol) und DIPEA (580 μΙ, 3.3 mmol) gegeben, und das Gemisch wurde 15 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-5-amino-4-[2- (trifluormethyl)phenyl]pentanoat (535 mg, 82% Reinheit, 1.38 mmol, Beispiel 279A), gelöst in DMF (3 ml) hinzugegeben, und das Gemisch wurde 18 h bei 60 °C gerührt. Anschließend wurde das Gemisch mit Ethylacetat und Wasser (jeweils 100 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (80 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung (100 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde mit- tels Flash-Säulenchromatographie (50 g Kieselgel Biotage Snap-Cartridge ultra, Cyclohexan/ Ethylacetat-Gradient 97:3— > 7:3, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 324 mg (100% Reinheit, 36% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.69 min; MS (ESIpos): m/z = 661/663 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 9.06 (t, 1 H), 8.03 (d, 1 H), 7.97 (dd, 1 H), 7.81-7.60 (m, 6H), 7.59-7.44 (m, 4H), 3.82-3.59 (m, 2H), 3.40-3.32 (m, 1 H), 2.26-1.84 (m, 4H), 1.34 (s, 9H).
Trennunq der Enantiomere:
Die Titelverbindung (228 mg) wurde in Isopropanol (6.7 ml) gelöst und mittels praparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 281A und 282A) [Säule: Daicel Chiralcel OZ-H, 5 μηη, 250 mm x 20 mm; Fluss: 20 ml/min; Detektion: 220 nm; Injektion: 0.17 ml; Eluent: 80% Heptan / 20% Isopropanol; Laufzeit 20 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet.
Beispiel 281A
(-)-ie f-Butyl-5-{[(6-brom-3-chlor-2-phenylchinolin-4-yl)carbonyl]amino}-4-[2-(trifluormethyl^ phenyl]pentanoat (Enantiomer 1)
Bei der in Beispiel 280A beschriebenen Enantiomerentrennung wurden 108 mg (100 % Reinheit, ee-Wert 97%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
Rt = 1 .52 min (chirale analytische HPLC; Daicel Chiralpak OZ-3, 3 μηη, 50 mm x 4.6 mm, Eluent Heptan / Isopropanol 80:20; Fluss 1 ml/min; Detektion 220 nM)
[a]D 20 = -8.9°, 589 nm, c = 0.45 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.68 min; MS (ESIpos): m/z = 661/663 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 9.06 (t, 1 H), 8.03 (d, 1 H), 7.97 (dd, 1 H), 7.80-7.60 (m, 6H), 7.59-7.52 (m, 3H), 7.49 (t, 1 H), 3.81 -3.60 (m, 2H), 3.4-3.3 (1 H, verdeckt), 2.26-1 .81 (m, 4H), 1 .37-1 .30 (m, 9H).
Beispiel 282A
(+)-ie/f-Butyl-5-{[(6-brom-3-chlor-2-phenylchinolin-4-yl)carbonyl]amino}-4-[2-(trifluormethyl)- phenyl]pentanoat (Enantiomer 2)
Bei der in Beispiel 280A beschriebenen Enantiomerentrennung wurden 102 mg (100 % Reinheit, ee-Wert 97%) der Titelverbindung als später eluierendes Enantiomer erhalten.
Rt = 2.57 min (chirale analytische HPLC; Daicel Chiralpak OZ-3, 3 μηη, 50 mm x 4.6 mm, Eluent Heptan / Isopropanol 80:20; Fluss 1 ml/min; Detektion 220 nM)
[a]D 20 = +10.0°, 589 nm, c = 0.37 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.68 min; MS (ESIpos): m/z = 661/663 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 9.06 (t, 1 H), 8.03 (d, 1 H), 7.97 (dd, 1 H), 7.80-7.60 (m, 6H), 7.59-7.51 (m, 3H), 7.49 (t, 1 H), 3.80-3.61 (m, 2H), 3.4-3.3 (m, 1 H, teilweise verdeckt), 2.25- 1 .82 (m, 4H), 1 .37-1 .30 (m, 9H).
Beispiel 283A
(+/-)-ie f-Butyl-4-(2-chlorphenyl)-5-{[(6-ethinyl-3-methyl-2-phenylch
pentanoat (Racemat)

Zu einem Gemisch aus 6-Ethinyl-3-methyl-2-phenylchinolin-4-carbonsäure (540 mg, 95% Reinheit, 1.77 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 109, Beispiel 97A) in DMF (8 ml) wurden HATU (810 mg, 2.13 mmol) und DIPEA (740 μΙ, 4.3 mmol) gegeben, und das Gemisch wurde 15 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-5-amino-4-(2-chlorphenyl)pentanoat (603 mg, 83% Reinheit, 1.77 mmol, Beispiel 121 A), gelöst in DMF (4 ml) hinzugegeben, und das Ge- misch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethyl- acetat und Wasser (jeweils 150 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (100 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung (200 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde mittels Flash-Säulenchromatographie (50 g Kieselgel Biotage Snap-Cartridge ultra, Cyclohexan/ Ethylacetat-Gradient 97:3— > 7:3, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 626 mg (100% Reinheit, 64% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.50 min; MS (ESIpos): m/z = 553 [M+H]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.83 (t, 1 H), 7.97 (d, 1 H), 7.72 (dd, 1 H), 7.7-7.4 (br. m, 1 H), 7.56-7.48 (m, 6H), 7.44 (d, 1 H), 7.37 (t, 1 H), 7.26 (t, 1 H), 4.42 (s, 1 H), 3.86-3.50 (m, 3H), 2.26-1.97 (m, 6H), 1.91 -1.72 (m, 1 H), 1.35 (s, 9H).
Trennung der Enantiomere:
Die Titelverbindung (481 mg) wurde in einem Gemisch aus Ethanol (3.5 ml) und Heptan (3.5 ml) gelöst und mittels präparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Bei- spiele 284A und 285A) [Säule: Daicel Chiralcel OX-H, 5 μηι, 250 mm x 20 mm; Fluss: 15 ml/min; Detektion: 220 nm; Injektion: 0.13 ml; Eluent: 80% Heptan / 20% Ethanol; Laufzeit 10 min, isok- ratisch]. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert.
Beispiel 284A
(-)-ie f-Butyl-4-(2-chlorphenyl)-5-{[(6-ethinyl-3-methyl-2-phenylchin
pentanoat (Enantiomer 1)
Bei der in Beispiel 283A beschriebenen Enantiomerentrennung wurden 205 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -17.9°, 589 nm, c = 0.44 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.48 min; MS (ESIpos): m/z = 553 [M+H]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.83 (t, 1 H), 7.97 (d, 1 H), 7.72 (dd, 1 H), 7.7-7.4 (br. m, 1 H), 7.56-7.47 (m, 6H), 7.44 (d, 1 H), 7.37 (t, 1 H), 7.26 (t, 1 H), 4.42 (s, 1 H), 3.84-3.51 (m, 3H), 2.23-1.94 (m, 6H), 1.89-1.71 (m, 1 H), 1.37 (s, 9H).
Beispiel 285A
(+)-ie/f-Butyl-4-(2-chlorphenyl)-5-{[(6-ethinyl-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- pentanoat (Enantiomer 2)
Bei der in Beispiel 283A beschriebenen Enantiomerentrennung wurden 217 mg (100% Reinheit, ee-Wert 99%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = +15.9°, 589 nm, c = 0.44 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.48 min; MS (ESIpos): m/z = 553 [M+H]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.83 (t, 1 H), 7.97 (d, 1 H), 7.72 (dd, 1 H), 7.7-7.4 (br. m, 1 H), 7.56-7.48 (m, 6H), 7.44 (d, 1 H), 7.37 (t, 1 H), 7.26 (t, 1 H), 4.41 (s, 1 H), 3.84-3.52 (m, 3H), 2.24-1.98 (m, 6H), 1.90-1.74 (m, 1 H), 1.37 (s, 9H).
Beispiel 286A
(+/-)-ie/f-Butyl-5-{[(6-brom-3-chlor-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlor-3,6-difluor- phenyl)pentanoat (Racemat)

Zu einem Gemisch aus 6-Brom-3-chlor-2-phenylchinolin-4-carbonsäure (500 mg, 1 .38 mmol, nicht reinheitskorrigiert, darstellbar gemäß WO 2016 146602 A1 , S. 70, Beispiel 37A) in DMF (6.0 ml) wurden HATU (629 mg, 1 .65 mmol) und DI PEA (580 μΙ, 3.3 mmol) gegeben, und das Gemisch wurde 15 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-5-amino-4-(2-chlor- 3,6-difluorphenyl)pentanoat (548 mg, 81 % Reinheit, 1 .38 mmol, Beispiel 128A), gelöst in DMF (3 ml) hinzugegeben, und das Gemisch wurde 3 h bei 60 °C bei RT gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethylacetat und Wasser (jeweils 100 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (80 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung (100 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde mittels Flash-Säulenchromatographie (50 g Kieselgel Biotage Snap-Cartridge ultra, Cyc- lohexan/ Ethylacetat-Gradient 97:3— > 7:3, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 296 mg (100% Reinheit, 32% d. Th.) der Titelverbindung erhalten. LC-MS (Methode 1 ): R
t = 2.67 min; MS (ESIpos): m/z = 663/665/667 [M+H]
+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 9.10 (t, 1 H), 8.02 (d, 1 H), 7.98 (dd, 1 H), 7.75-7.63 (m, 3H), 7.59-7.50 (m, 3H), 7.41 (td, 1 H), 7.30 (td, 1 H), 3.91 -3.63 (m, 3H), 2.23-1 .86 (m, 4H), 1 .36 (s, 9H).
Trennung der Enantiomere:
Die Titelverbindung (215 mg) wurde in Ethanol (6 ml) gelöst und mittels präparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 287A und 288A) [Säule: Daicel Chi- ralpak AD-H, 5 μηη, 250 mm x 30 mm; Fluss: 40 ml/min; Detektion: 240 nm; Temperatur: 30 °C; Injektion: 0.35 ml; Eluent: 80% Heptan / 20% Isopropanol; isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand in Acetonitril/Wasser lyophilisiert.
Beispiel 287A
(-)-ie/f-Butyl-5-{[(6-brom-3-chlor-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlor-3,6-difluor- phenyl)pentanoat (Enantiomer 1)
Bei der in Beispiel 286A beschriebenen Enantiomerentrennung wurden 66 mg (96% Reinheit, ee-Wert 99%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
Rt = 1 .28 min (chirale analytische HPLC; Daicel Chiralpak AD 3 μηη, 50 mm x 4.6 mm, Eluent Heptan / Isopropanol 80:20; Fluss 1 ml/min; Detektion 220 nM)
[a]D 20 = -27.1 °, 589 nm, c = 0.38 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.64 min; MS (ESIpos): m/z = 663/665/667 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 9.10 (t, 1 H), 8.02 (d, 1 H), 7.98 (dd, 1 H), 7.77-7.61 (m, 3H), 7.59-7.50 (m, 3H), 7.41 (td, 1 H), 7.30 (td, 1 H), 3.91 -3.65 (m, 3H), 2.22-2.04 (m, 3H), 1 .98 (br. s, 1 H), 1.36 (s, 9H).
Beispiel 288A
(+)-ie f-Butyl-5-{[(6-brom-3-chlor-2-phenylchinolin-4-yl)carbonyl]ami
phenyl)pentanoat (Enantiomer 2)
Bei der in Beispiel 286A beschriebenen Enantiomerentrennung wurden 64 mg (92% Reinheit, ee-Wert 98%) der Titelverbindung als später eluierendes Enantiomer erhalten.
Rt = 1 .42 min (chirale analytische HPLC; Daicel Chiralpak AD 3 μηη, 50 mm x 4.6 mm, Eluent Heptan / Isopropanol 80:20; Fluss 1 ml/min; Detektion 220 nM)
[a]D 20 = +24.9°, 589 nm, c = 0.38 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.65 min; MS (ESIpos): m/z = 663/665/667 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 9.10 (t, 1 H), 8.02 (d, 1 H), 7.98 (dd, 1 H), 7.75-7.62 (m, 3H), 7.59-7.50 (m, 3H), 7.41 (td, 1 H), 7.30 (td, 1 H), 4.17-3.64 (m, 3H, teilweise verdeckt), 2.24- 2.03 (m, 3H), 1 .98 (br. s, 1 H), 1 .36 (s, 9H).
Beispiel 289A
(+/-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(pyridin-2-yl)- pentanoat (Racemat)
6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (213 mg, 622 μηηοΙ, darstellbar gemäß WO 2016 146602 A1 , S. 70, Beispiel 37A) wurde in DMF (7 ml) aufgenommen und mit HATU (284 mg, 747 μηιοΙ) versetzt. Nach 5 min Rühren bei RT unter Stickstoff wurde DIPEA (330 μΙ, 1 .9 mmol) hinzugegeben, und das Gemisch wurde für 30 min bei RT weitergerührt. Anschließend wurde (+/-)-ie/f-Butyl-5-amino-4-(pyridin-2-yl)pentanoat (187 mg, 747 μηηοΙ, Beispiel 146A, nicht reinheitskorrigiert) in 20 ml DMF gelöst und zum Reaktionsgemisch gegeben. Es wurde 2 h
bei RT nachgerührt. Das Reaktionsgemisch wurde direkt (ohne weitere Aufarbeitung) chromatographisch aufgereinigt (Biotage Isolera Four, LiChroprep RP-18 (40-63 μηι); Gradient 30%— > 90% Acetonitril in 0.1 % wässriger Ammoniaklösung). Es wurden 100 mg (80% Reinheit, 1 % d. Th. über 3 Stufen) der Titelverbindung erhalten.
LC-MS (Methode 6): Rt = 1.43 min; MS (ESIpos): m/z = 574/576 [M+H]+ Beispiel 290A
(+/-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-[2-fluor-6-(trifluor- methyl)phenyl]pentanoat (Racemat)

Zu einer Lösung von 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (939 mg, 2.74 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (32 ml) wurden HATU (1.25 g, 3.29 mmol) und DIPEA (1.1 ml, 6.6 mmol) hinzugegeben. Das Gemisch wurde 15 min bei RT gerührt. Eine Lösung von (+/-)-ierf-Butyl-5-amino-4-[2-fluor-6-(trifluormethyl)phenyl]pentanoat (1.00 g, 92% Reinheit, 2.74 mmol, Beispiel 147A) in wenig DMF wurde hinzugefügt und die Re- aktionsmischung 1 h bei 60 °C, dann noch 30 min bei RT gerührt. Das Gemisch wurde mit je 200 ml Wasser und Essigsaureethylester versetzt. Die Phasen wurden getrennt. Die organische Phase wurde mit 200 ml Wasser, dann mit 200 ml einer gesättigten Natriumchlorid Lösung gewaschen, über Natriumsulfat getrocknet und in Vakuum eingeengt. Der Rückstand wurde in wenig DMSO gelöst und mittels präparativer HPLC (Methode 28) aufgereinigt. Die geeigneten Frak- tionen wurden zusammen eingeengt und im Vakuum getrocknet. Es wurde 1.29 g (93% Reinheit, 66% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.64 min; MS (ESIpos): m/z = 659/661 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.95 (t, 1 H), 7.95 (d, 1 H), 7.85 (dd, 1 H), 7.80-7.45 (m, 9H), 3.93 (br. s, 1 H), 3.74 (br. s, 1 H), 3.33 (1 H, verdeckt, tentativ), 2.28-2.00 (m, 7H), 1.34 (s, 9H).
Trennunq der Enantiomere:
Das so erhaltene Racemat (1 .2 g) wurde in 140 ml Acetonitril und wenig Methanol gelöst und portionsweise mittels praparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 291A und 292A) [Methode: Säule Daicel Chiralpak AD-H 5 μηι, 250 mm x 20 mm; Eluent Kohlendioxid / Isopropanol 80:20 isokratisch; Fluss 80 ml/min; Temperatur 40 °C; Detek- tion 210 nM]. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde in Ace- tonitril/Wasser lyophilisiert.
Beispiel 291A
ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-[2-fluor-6-(trifluormethyl)- phenyl]pentanoat (Enantiomer 1)
Bei der in Beispiel 290A beschriebenen Enantiomerentrennung wurden 444 mg (97% Reinheit, ee-Wert 100%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
Rt = 1 .41 min (chirale analytische HPLC; Daicel Chiralpak AD 3 μηι, 100 mm x 4 mm, Eluent Kohlendioxid / Isopropanol 80:20; Fluss 3 ml/min; Temperatur 40 °C; Detektion 210 nM)
LC-MS (Methode 1 ): Rt = 2.62 min; MS (ESIpos): m/z = 659/661 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.41 ), 1 .030 (1 .89), 1 .046 (1 .90), 1 .336 (16.00), 2.086 (0.51 ), 2.107 (0.49), 2.125 (0.65), 2.152 (0.53), 2.167 (0.49), 7.507 (0.85), 7.513 (0.68), 7.525 (1 .40), 7.538 (1 .61 ), 7.555 (0.43), 7.562 (0.47), 7.573 (0.50), 7.595 (0.46), 7.861 (0.58), 7.867 (0.57), 7.939 (1 .06), 7.961 (0.69). Beispiel 292A
ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-[2-fluor-6- (trifluormethyl)phenyl]pentanoat (Enantiomer 2)
Bei der in Beispiel 290A beschriebenen Enantiomerentrennung wurden 416 mg (97% Reinheit, ee-Wert 100%) der Titelverbindung als später eluierendes Enantiomer erhalten.
Rt = 2.01 min (chirale analytische HPLC; Daicel Chiralpak AD 3 μηι, 100 mm x 4 mm, Eluent Kohlendioxid / Isopropanol 80:20; Fluss 3 ml/min; Temperatur 40 °C; Detektion 210 nM)
LC-MS (Methode 1 ): Rt = 2.62 min; MS (ESIpos): m/z = 659 [M+H]+
1 H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.56), 0.008 (0.56), 1 .030 (1 .54), 1 .045 (1 .55), 1 .336 (16.00), 2.085 (0.51 ), 2.107 (0.48), 2.124 (0.65), 2.152 (0.53), 2.167 (0.49), 7.506 (0.83), 7.513 (0.67), 7.524 (1 .38), 7.537 (1 .60), 7.555 (0.43), 7.562 (0.46), 7.573 (0.51 ), 7.594 (0.46), 7.861 (0.58), 7.867 (0.58), 7.938 (1 .06), 7.961 (0.68).
Beispiel 293A
(+/-)-ie f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]a
phenyl)pentanoat (Racemat)

Zu einer Lösung aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (593 mg, 1.73 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (20 ml) wurden HATU (791 mg, 2.08 mmol) und DIPEA (720 μΙ, 4.2 mmol) hinzugegeben. Das Gemisch wurde 15 min bei RT gerührt. Eine Lösung von (+/-)-ie/f-Butyl-5-amino-4-(2,3,5,6-tetrafluorphenyl)pentanoat (586 mg, 95% Reinheit, 1.73 mmol, Beispiel 148A) in wenig DMF wurde hinzugefügt und die Reaktionsmischung 1 h bei 60 °C, dann weiter über Nacht bei RT gerührt. Das Gemisch wurde mit je 100 ml Wasser und Ethylacetat versetzt. Die Phasen wurden getrennt. Die wäßrige Phase wurde mit 100 ml Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit 200 ml einer gesättigten Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet und im Vakuum eingeengt. Der Rückstand wurde in wenig DMSO gelöst und mittels präparativer HPLC (Methode 28) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurde 700 mg (100% Reinheit, 63% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.56 min; MS (ESIpos): m/z = 645/647 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.52), 0.008 (0.56), 1.366 (16.00), 2.073 (0.62), 2.156 (1.15), 2.178 (0.49), 2.196 (0.50), 2.212 (0.40), 2.228 (0.44), 3.789 (0.44), 3.805 (0.64), 7.506 (0.79), 7.513 (0.77), 7.524 (2.69), 7.535 (1.15), 7.539 (1.01 ), 7.849 (0.47), 7.854 (0.43), 7.871 (0.67), 7.877 (0.67), 7.941 (1.24), 7.964 (0.76), 8.981 (0.50).
Trennung der Enantiomere:
Die Titelverbindung (658 mg) wurde in Methanol (30 ml) gelöst und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 294A und 295A) [Säule: Säule Daicel Chiralpak AD-H 5 μηη, 250 mm x 20 mm; Eluent Kohlendioxid / Isopropanol 80:20 isokra- tisch; Fluss 80 ml/min; Temperatur 40 °C; Detektion 210 nM; isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde in Acetonitril/Wasser lyophilisiert.
Beispiel 294A
ie f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]am
phenyl)pentanoat (Enantiomer 1)
Bei der in Beispiel 293A beschriebenen Enantiomerentrennung wurden 266 mg (100% Reinheit, ee-Wert 100%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
Rt = 1 .37 min (chirale analytische HPLC; Daicel Chiralpak AD 3 μηη, 100 mm x 4 mm, Eluent Kohlendioxid / Isopropanol 80:20; Fluss 3 ml/min; Temperatur 40 °C; Detektion 210 nM)
Beispiel 295A
ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl )carbonyl]amino}-4-(2, 3,5,6- tetrafluorphenyl)pentanoat (Enantiomer 2)
Bei der in Beispiel 293A beschriebenen Enantiomerentrennung wurden 275 mg (99% Reinheit, ee-Wert 99%) der Titelverbindung als später eluierendes Enantiomer erhalten.
Rt = 1 .67 min (chirale analytische HPLC; Daicel Chiralpak AD 3 μηη, 100 mm x 4 mm, Eluent Kohlendioxid / Isopropanol 80:20; Fluss 3 ml/min; Temperatur 40 °C; Detektion 210 nM) Beispiel 296A
(+/-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2,3,5-trifluor- phenyl)pentanoat (Racemat)
Zu einer Lösung von 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (217 mg, 633 μηηοΙ, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (8.6 ml) wurden HATU (289 mg, 760 μηηοΙ) und DI PEA (260 μΙ, 1.5 mmol) hinzugegeben. Das Gemisch wurde 15 min bei RT gerührt. Eine Lösung von (+/-)-ie/f-Butyl-5-amino-4-(2,3,5-trifluorphenyl)pentanoat (200 mg, 96% Reinheit, 633 μηηοΙ, Beispiel 149A) in wenig DMF wurde hinzugefügt und die Reaktionsmischung 3
h bei 60 °C gerührt. Das Gemisch wurde mit Wasser und Ethylacetat (jeweils 100 ml) versetzt. Die Phasen wurden getrennt. Die wässrige Phase wurde mit Ethylacetat (50 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter Natriumchlorid Lösung (100 ml) gewaschen, über Natriumsulfat getrocknet und im Vakuum eingeengt. Der Rückstand wurde in wenig Dichlor- methan gelöst und mittels Flash-Chromatographie (Biotage, 50 g Kieselgel, Cyclohexan / Ethyl- acetat-Gradient 100:0— > 80:20) gereinigt. Die laut DC sauberen Fraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 1 15 mg (100% Reinheit, 29% d. Th., siehe Analytik) einer ersten Charge der Titelverbindung erhalten. Eine erhaltene, unsaubere Fraktion wurde eingeengt, und der Rückstand wurde mittels präparativer HPLC (Methode 32) gereinigt. Die verei- nigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 25 mg (100% Reinheit, 6% d. Th.) einer zweiten Charge der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.56 min; MS (ESIpos): m/z = 627/629 [M+H]+
1 H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1 .371 (16.00), 1 .398 (9.99), 1 .826 (0.18), 2.038 (0.21 ), 2.127 (0.89), 3.413 (0.18), 3.714 (0.27), 7.233 (0.22), 7.413 (0.17), 7.504 (0.77), 7.512 (0.75), 7.523 (2.51 ), 7.839 (0.38), 7.844 (0.35), 7.861 (0.58), 7.866 (0.56), 7.935 (1 .13), 7.957 (0.72), 8.889 (0.46).
1 H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.89 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.66 (br. s, 1 H), 7.55-7.48 (m, 5H), 7.46-7.34 (m, 1 H), 7.30-7.17 (m, 1 H), 3.86-3.63 (m, 2H), 3.48-3.37 (m, 1 H), 2.23-1 .93 (m, 6H), 1 .90-1.75 (m, 1 H), 1 .37 (s, 9H).
Trennung der Enantiomere:
Die Titelverbindung (108 mg) wurde in Methanol (12 ml) gelöst und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 297A und 298A) [Säule: Säule Daicel Chiralpak AD-H 5 μηη, 250 mm x 20 mm; Eluent Kohlendioxid / Isopropanol 80:20 isokra- tisch; Fluss 80 ml/min; Temperatur 40 °C; Detektion 210 nM; isokratisch]. Die vereinigten Ziel- fraktionen wurden eingeengt, und der Rückstand wurde in Acetonitril/Wasser lyophilisiert.
Beispiel 297A
ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2,3,5-trifluorphenyl)- pentanoat (Enantiomer 1)
Bei der in Beispiel 296A beschriebenen Enantiomerentrennung wurden 39 mg (97% Reinheit, ee-Wert 100%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
Rt = 0.82 min (chirale analytische HPLC; Daicel Chiralpak AD 3 μηη, 100 mm x 4 mm, Eluent Kohlendioxid / Isopropanol 75:25; Fluss 3 ml/min; Temperatur 40 °C; Detektion 210 nM)
Beispiel 298A
ie f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]am
pentanoat (Enantiomer 2)
Bei der in Beispiel 296A beschriebenen Enantiomerentrennung wurden 41 mg (99% Reinheit, ee-Wert 99%) der Titelverbindung als später eluierendes Enantiomer erhalten.
Rt = 1 .03 min (chirale analytische HPLC; Daicel Chiralpak AD 3 μηη, 100 mm x 4 mm, Eluent Kohlendioxid / Isopropanol 75:25; Fluss 3 ml/min; Temperatur 40 °C; Detektion 210 nM)
Beispiel 299A
(+/-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2,3,6-trichlor- phenyl)pentanoat (Racemat)
Zu einer Lösung von 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (604 mg, 1 .77 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (20 ml) wurden HATU (806 mg, 2.12 mmol) und DI PEA (740 μΙ, 4.2 mmol) hinzugegeben. Das Gemisch wurde 15 min bei RT gerührt. Eine Lösung von (+/-)-ie/f-Butyl-5-amino-4-(2,3,6-trichlorphenyl)pentanoat (700 mg, 89% Reinheit, 1 .77 mmol, Beispiel 150A) in wenig DMF wurde hinzugefügt und die Reaktionsmischung 1 h bei 60 °C, gefolgt von 30 min bei RT gerührt. Das Gemisch wurde mit Wasser und Ethylacetat (jeweils 100 ml) versetzt. Die Phasen wurden getrennt. Die organische Phase wurde mit Wasser (200 ml) und dann mit gesättigter wässriger Natriumchlorid-Lösung (200 ml) gewa- sehen, über Natriumsulfat getrocknet und im Vakuum eingeengt. Der Rückstand wurde in wenig DMSO gelöst und mittels präparativer HPLC (Methode 28) aufgereinigt. Die vereinigten Zielfraktionen wurden im Vakuum eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurde 745 mg (100% Reinheit, 62% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.78 min; MS (ESIpos): m/z = 675/677/679 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1 .61 ), 0.008 (1 .78), 1 .331 (0.43), 1 .349 (15.88), 1 .353 (16.00), 2.073 (1 .05), 2.092 (0.49), 2.106 (0.54), 2.120 (0.68), 2.133 (1 .1 1 ), 2.146 (1 .43), 2.163 (1 .36), 2.179 (3.06), 2.523 (0.43), 4.067 (0.43), 4.090 (0.45), 7.472 (0.45), 7.494 (0.94), 7.504 (1 .43), 7.509 (1 .14), 7.512 (1 .16), 7.528 (4.20), 7.550 (1 .61 ), 7.582 (0.54), 7.591 (0.58), 7.604 (0.41 ), 7.841 (0.50), 7.847 (0.60), 7.864 (0.75), 7.869 (0.95), 7.876 (0.57), 7.935 (1 .21 ), 7.942 (1 .1 1 ), 7.958 (0.77), 7.964 (0.67), 8.920 (0.47), 8.935 (0.54).
Trennung der Enantiomere:
Die Titelverbindung (604 mg) wurde in Isopropanol (12 ml) gelöst und mittels praparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 300A und 301A) [Säule: Daicel Chiralpak I E, 5 μηη, 250 mm x 20 mm; Eluent Heptan / Isopropanol 80:20 isokratisch; Fluss 15 ml/min; Temperatur 40 °C; Detektion 220 nM, Laufzeit 18 min]. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde jeweils in Aceton itril/Wasser lyophilisiert.
Beispiel 300A
ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2,3,6-trichlorphenyl)- pentanoat (Enantiomer 1)
Bei der in Beispiel 299A beschriebenen Enantiomerentrennung wurden 321 mg (93% Reinheit, ee-Wert 96%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
Rt = 7.19 min (chirale analytische HPLC; Daicel Chiralpak I E 5 μηη, 250 mm x 4.6 mm, Eluent Heptan / 0.2% Diethylamin in Isopropanol 50:50, isokratisch; Fluss 1 ml/min; Temperatur 40 °C; Detektion 210 nM)
Beispiel 301
ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2,3,6-trichlorphenyl)- pentanoat (Enantiomer 1)
Bei der in Beispiel 299A beschriebenen Enantiomerentrennung wurden 291 mg (99% Reinheit, ee-Wert 100%) der Titelverbindung als später eluierendes Enantiomer erhalten.
Rt = 8.98 min (chirale analytische HPLC; Daicel Chiralpak I E 5 μηη, 250 mm x 4.6 mm, Eluent Heptan / 0.2% Diethylamin in Isopropanol 50:50, isokratisch; Fluss 1 ml/min; Temperatur 40 °C; Detektion 210 nM)
Beispiel 302A
(+/-)-ie/f-Butyl-6-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-5-(2-chlorphenyl)- hexanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (1.03 g, 3.00 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (12 ml) wurden HATU (1.37 g, 3.60 mmol) und DIPEA (1.3 ml, 7.2 mmol) gegeben und das Gemisch wurde 15 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-6-amino-5-(2-chlorphenyl)hexanoat (893 mg, 3.00 mmol, Beispiel 151A), gelöst in DMF (9 ml) hinzugegeben, und das Gemisch wurde 3 h bei 60 °C gerührt. Anschließend wurde das Gemisch mit Ethylacetat und Wasser (jeweils 100 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase zweimal mit Ethylacetat (80 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natri- umchlorid-Lösung (100 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge ultra, Cyclohexan / Ethylacetat 98:2— > 8:2, Iso- lera One) gereinigt. Es wurden 1.19 g (93% Reinheit, 59% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.66 min; MS (ESIpos): m/z = 621/623 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.84 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.72 (br. s, 1 H), 7.56-7.41 (m, 7H), 7.41-7.17 (m, 2H), 3.70 (br. s, 2H), 3.62-3.52 (m, 1 H), 2.25-2.05 (m, 5H), 1.86-1.70 (m, 1 H), 1.69-1.55 (m, 1 H), 1.54-1.30 (m, 2H), 1.35 (s, 9H).
Trennung der Enantiomere:
Die Titelverbindung (1.06 g) wurde in Methanol (50 ml) gelöst und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 303A und 304A) [Säule: Daicel Chi- ralcel OJ-H, 5 μηη, 250 mm x 30 mm; Fluss: 100 ml/min; Detektion: 210 nm; Temperatur: 38 °C; Injektion: 1.0 ml; Eluent: 82% Kohlendioxid / 18% Methanol -> 67% Kohlendioxid / 33% Methanol; Laufzeit 1 1 min]. Die vereinigten Zielfraktionen wurden eingeengt, und der jeweilige Rückstand wurde im Vakuum getrocknet. Beispiel 303A
ie/f-Butyl-6-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-5-(2-chlorphenyl)hexanoat (Enantiomer 1)
Bei der in Beispiel 302A beschriebenen Enantiomerentrennung wurden 480 mg (100% Reinheit, ee-Wert 100%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
Rt = 0.90 min (chirale analytische HPLC; Daicel Chiralpak OJ-3, 3 μηη, 100 mm x 4.6 mm, Eluent Kohlendioxid / Methanol 8:2, isokratisch; Fluss 3 ml/min; Druck 130 bar, Temperatur 40 °C; De- tektion 210 nM)
LC-MS (Methode 1): Rt = 2.67 min; MS (ESIpos): m/z = 621/623 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.149 (0.03), 0.146 (0.03), 1.194 (0.07), 1.320 (0.16), 1.354 (16.00), 1.398 (0.17), 1.421 (0.14), 1.438 (0.18), 1.448 (0.18), 1.463 (0.20), 1.480 (0.15), 1.496 (0.09), 1.511 (0.11), 1.576 (0.08), 1.609 (0.19), 1.622 (0.17), 1.634 (0.20), 1.732 (0.19), 1.745 (0.20), 1.759 (0.20), 1.792 (0.11), 2.145 (0.99), 2.165 (0.66), 2.171 (0.59), 2.183 (0.80), 2.189 (0.76), 2.201 (0.39), 2.207 (0.38), 2.229 (0.08), 2.328 (0.04), 2.366 (0.04), 2.670 (0.04), 2.710 (0.03), 3.581 (0.25), 3.597 (0.22), 3.701 (0.39), 7.236 (0.19), 7.254 (0.40), 7.273 (0.28), 7.298 (0.08), 7.317 (0.06), 7.350 (0.24), 7.369 (0.43), 7.387 (0.23), 7.433 (0.65), 7.453 (0.54), 7.478 (0.65), 7.499 (1.20), 7.507 (0.82), 7.519 (2.58), 7.719 (0.09), 7.833 (0.38), 7.838 (0.36), 7.856 (0.58), 7.860 (0.57), 7.930 (1.03), 7.953 (0.66), 8.832 (0.24), 8.846 (0.48), 8.859 (0.24).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.85 (t, 1H), 7.94 (d, 1H), 7.85 (dd, 1H), 7.72 (br. s, 1H), 7.56-7.42 (m, 7H), 7.40-7.20 (m, 2H), 3.70 (br. s, 2H), 3.63-3.50 (m, 1H), 2.24-2.08 (m, 5H), 1.88-1.71 (m, 1H), 1.69-1.54 (m, 1H), 1.53-1.30 (m, 2H), 1.35 (s, 9H).
Beispiel 304A
ie/f-Butyl-6-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-5-(2-chlorphenyl)hexanoat (Enantiomer 2)
Bei der in Beispiel 302A beschriebenen Enantiomerentrennung wurden 438 mg (100% Reinheit, ee-Wert 100%) der Titelverbindung als später eluierendes Enantiomer erhalten.
Rt = 3.05 min (chirale analytische HPLC; Daicel Chiralpak OJ-3, 3 μηη, 100 mm x 4.6 mm, Eluent Kohlendioxid / Methanol 8:2, isokratisch; Fluss 3 ml/min; Druck 130 bar, Temperatur 40 °C; De- tektion 210 nM)
LC-MS (Methode 1): Rt = 2.68 min; MS (ESIpos): m/z = 621/623 [M+H]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.194 (0.07), 1.354 (16.00), 1.420 (0.16), 1.462 (0.21), 1.480 (0.16), 1.510 (0.11), 1.609 (0.19), 1.633 (0.21), 1.731 (0.19), 1.757 (0.21), 2.144 (1.03), 2.164 (0.69), 2.182 (0.84), 2.188 (0.79), 2.200 (0.42), 2.327 (0.06), 2.669 (0.05), 3.580 (0.25), 3.701 (0.42), 7.235 (0.19), 7.254 (0.41), 7.272 (0.29), 7.350 (0.25), 7.368 (0.45), 7.386 (0.25), 7.433 (0.65), 7.452 (0.55), 7.479 (0.68), 7.498 (1.24), 7.507 (0.91), 7.518 (2.62), 7.714 (0.10), 7.833 (0.40), 7.838 (0.38), 7.855 (0.62), 7.860 (0.60), 7.930 (1.04), 7.952 (0.67), 8.829 (0.26), 8.844 (0.49), 8.857 (0.25).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.84 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.71 (br. s, 1 H), 7.57-7.42 (m, 7H), 7.37 (t, 1 H), 7.26 (t, 1 H), 3.70 (br. s, 2H), 3.63-3.51 (m, 1 H), 2.26-2.06 (m, 5H), 1.85-1.70 (m, 1 H), 1.69-1.55 (m, 1 H), 1.53-1.30 (m, 2H), 1.35 (s, 9H).
Beispiel 305A
(+/-)-ie f-Butyl-6-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]am
methylhexanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (895 mg, 2.62 mmol, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (10 ml) wurden HATU (1.19 g, 3.14 mmol) und DIPEA (1.1 ml, 6.3 mmol) gegeben und das Gemisch wurde 15 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-6-amino-5-(2-chlorphenyl)-5-methylhexanoat (816 mg, „2.62 mmol", Beispiel 152A, nicht reinheitskorrigiert, im Gemisch mit (+/-)-ierf-Butyl-6-amino-5- methyl-5-phenylhexanoat), gelöst in DMF (8 ml) hinzugegeben, und das Gemisch wurde 3 h bei 60 °C, gefolgt von 18 h bei RT gerührt. Anschließend wurde das Gemisch mit Ethylacetat und Wasser (jeweils 150 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase zweimal mit Ethylacetat (80 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung (200 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan / Ethylacetat 97:3 — > 7:3, Isolera One) gereinigt. Nach Einengen und Trocknen der Rückstände im Vakuum wurden zwei Mischfraktionen erhalten, in denen jeweils die Zielverbindung im Gemisch mit dem dechlorierten Produkt (ie/f-Butyl-6-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-5- methyl-5-phenylhexanoat) enthalten war. Die erste Mischfraktion (866 mg) wurde für die betreffende Folgereaktion (siehe Beispiel 240) verwendet. Die zweite Mischfraktion (386 mg) wurde in ei- nem Gemisch aus DMSO, Acetonitril und THF gelöst und mittels präparativer HPLC (Methode 15) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und die Rückstände wurden lyophilisiert. Es wurden 161 mg (100% Reinheit) der Titelverbindung erhalten. Außerdem wurden 148 mg
(97% Reinheit) des de-chlorierten Produktes (ierf-Butyl-6-{[(6-brom-3-methyl-2-phenylchinolin-4- yl)carbonyl]amino}-5-methyl-5-phenylhexanoat, siehe Beispiel 306A) erhalten.
LC-MS (Methode 1 ): Rt = 2.76 min; MS (ESIpos): m/z = 635/637 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.074 (0.18), 1.359 (16.00), 1.526 (2.00), 1.618 (0.24), 2.1 18 (0.75), 2.136 (1.16), 2.154 (0.58), 2.394 (0.20), 3.672 (0.21 ), 3.693 (0.22), 4.298 (0.36), 4.316 (0.38), 4.332 (0.35), 4.349 (0.32), 7.260 (0.35), 7.279 (0.32), 7.309 (0.41 ), 7.327 (0.20), 7.41 1 (0.44), 7.430 (0.78), 7.446 (0.42), 7.498 (0.93), 7.506 (0.92), 7.517 (2.01 ), 7.828 (0.37), 7.850 (0.53), 7.855 (0.48), 7.924 (0.98), 7.946 (0.60), 8.599 (0.40).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.60 (t, 1 H), 7.93 (d, 1 H), 7.84 (dd, 1 H), 7.69 (br. s, 1 H), 7.56-7.47 (m, 5H), 7.46-7.38 (m, 2H), 7.34-7.23 (m, 2H), 4.32 (dd, 1 H), 3.68 (dd, 1 H), 2.42-2.32 (m, 1 H), 2.22-2.02 (m, 5H), 1.62 (td, 1 H), 1.53 (s, 3H), 1.4-1.3 (m, 1 H, teilweise verdeckt), 1.39 (s, 3H), 1.36 (s, 9H), 1.14-0.99 (m, 1 H).
Beispiel 306A
(+/-)-ie/f-Butyl-6-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-5-methyl-5-phenyl- hexanoat (Racemat)
Wie in Beispiel 305A beschrieben, wurden 148 mg (97% Reinheit) der Titelverbindung erhalten. LC-MS (Methode 1 ): Rt = 2.70 min; MS (ESIpos): m/z = 601/603 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.162 (0.17), 1.351 (16.00), 1.388 (3.98), 1.599 (0.22), 1.631 (0.15), 1.762 (0.14), 1.784 (0.20), 2.099 (0.70), 2.1 18 (1.20), 2.136 (0.63), 2.365 (0.12), 3.540 (0.21 ), 3.562 (0.26), 3.727 (0.45), 3.843 (0.28), 3.860 (0.28), 3.876 (0.23), 3.893 (0.21 ), 7.215 (0.39), 7.233 (0.26), 7.326 (0.43), 7.345 (0.86), 7.364 (0.54), 7.405 (1.12), 7.424 (0.66), 7.499 (0.78), 7.506 (0.68), 7.517 (1.61 ), 7.525 (1.49), 7.827 (0.35), 7.850 (0.51 ), 7.855 (0.50), 7.927 (0.97), 7.950 (0.62), 8.608 (0.38).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.61 (t, 1 H), 7.94 (d, 1 H), 7.84 (dd, 1 H), 7.67 (br. s, 1 H), 7.56-7.46 (m, 5H), 7.44-7.39 (m, 2H), 7.35 (t, 2H), 7.22 (t, 1 H), 3.87 (dd, 1 H), 3.55 (dd, 1 H), 2.22-2.02 (m, 5H), 1.79 (td, 1 H), 1.60 (td, 1 H), 1.4-1.3 (m, 1 H, teilweise verdeckt), 1.39 (s, 3H), 1.35 (s, 9H), 1.23-1.07 (m, 1 H). Beispiel 307A
2-(Brommethyl)-1-(difluormethoxy)-3-fluorbenzol
Zu einer Lösung aus [2-(Difluormethoxy)-6-fluorphenyl]methanol (9.90 g, 51.5 mmol, Beispiel 3A) in Dichlormethan (60 ml) wurde bei -15 °C eine Lösung aus Phosphortribromid (1.6 ml, 16 mmol) in Dichlormethan (20 ml) unter Rühren tropfenweise gegeben. Anschließend wurde das Kältebad entfernt, und das Gemisch wurde weitere 2 h gerührt. Danach wurde das Gemisch langsam mit gesättigter Natriumhydrogencarbonat-Lösung, Wasser und Dichlormethan (jeweils 100 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die organische Phase mit gesättigter Natriumchlorid-Lösung (200 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde kurz im Vakuum getrocknet. Es wurden 9.60 g (95% Reinheit, 69% d. Th.) der Titelverbindung erhalten.
GC-MS (Methode 12): Rt = 3.38 min, MS (Elpos): m/z = 254/256 [M]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.238 (0.57), 1.255 (1.12), 1.273 (0.58), 4.610 (15.58), 4.614 (16.00), 7.105 (3.32), 7.126 (3.80), 7.161 (2.02), 7.184 (8.39), 7.205 (2.57), 7.366 (8.46), 7.470 (2.03), 7.487 (2.27), 7.491 (3.74), 7.508 (3.68), 7.512 (2.00), 7.529 (1.66), 7.549 (4.14).
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 7.60-7.03 (m, 4H), 4.61 (d, 2H). Beispiel 308A
[2-(Difluormethoxy)-6-fluorphenyl]acetonitril
Zu einer Lösung aus 2-(Brommethyl)-1-(difluormethoxy)-3-fluorbenzol (9.60 g, 37.6 mmol, Beispiel 307A) in Dichlormethan (60 ml) wurden unter Rühren Wasser (60 ml) und Tetrabutylam-
moniumbromid (1.21 g, 3.76 mmol) gegeben. Anschließend wurde eine Lösung aus Kaliumcya- nid (7.35 g, 1 13 mmol) in Wasser (120 ml) hinzugegeben, und das Gemisch wurde 2.5 h bei RT gerührt. Anschließend wurden die Phasen getrennt, und die organische Phase wurde zweimal mit gesättigter Natriumhydrogencarbonat-Lösung (jeweils 100 ml) gewaschen, über Natriumsul- fat getrocknet, filtriert und eingeengt, und der Rückstand wurde kurz im Vakuum getrocknet. Es wurden 7.26 g (98% Reinheit, 94% d. Th.) der Titelverbindung erhalten.
GC-MS (Methode 12): Rt = 3.70 min, MS (Elpos): m/z = 201 [M]+
LC-MS (Methode 1 ): Rt = 1.56 min
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.937 (0.68), 3.164 (0.78), 3.177 (0.77), 3.952 (16.00), 7.152 (3.20), 7.171 (6.16), 7.215 (2.00), 7.237 (3.89), 7.259 (2.35), 7.353 (6.55), 7.489 (1.60), 7.509 (2.98), 7.526 (3.04), 7.531 (1.93), 7.535 (3.48), 7.547 (1.32).
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 7.58-7.43 (m, 1 H), 7.39-7.05 (m, 3H), 3.95 (s, 2H). Beispiel 309A
(+/-)-ie/f-Butyl-4-cyan-4-[2-(difluormethoxy)-6-fluorphenyl]butanoat (Racemat)
Zu einer Lösung aus [2-(Difluormethoxy)-6-fluorphenyl]acetonitril (7.24 g, 36.0 mmol, Beispiel 308A) in THF (30 ml) unter Argon wurde bei ca. -70 bis -60 °C langsam unter Rühren eine 2 M Lösung aus LDA in THF (22 ml, 43 mmol) gegeben. Das Gemisch wurde auf 0 °C kommen gelassen und nach 15 min wieder auf -70 °C abgekühlt. Anschließend wurde eine Lösung aus ie f-Butyl-3-brompropanoat (6.8 ml, 43 mmol) in THF (15 ml) langsam unter Rühren bei ca. -70 bis -60 °C hinzu getropft. Das Gemisch wurde über Nacht weiter gerührt, wobei das Kältebad (Trockeneis/Isopropanol) langsam auf RT kommen gelassen wurde. Anschließend wurde das Gemisch bei ca. 0 °C langsam mit Wasser und Ethylacetat (jeweils 100 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (100 ml) ex- trahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter Natriumchlorid- Lösung (150 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (400 g Kieselgel, Cyclohexan/Ethylacetat-Gradient 10:1 ) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 7.1 1 g (74% Reinheit, 45% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.10 min; MS (ESIpos): m/z = 330 [M+H]+
Beispiel 310A
(+/-)-ie/f-Butyl-5-amino-4-[2-(difluormethoxy)-6-fluorphenyl]pentanoat (Racemat)
Eine Lösung aus (+/-)-ie/f-Butyl-4-cyan-4-[2-(difluormethoxy)-6-fluorphenyl]butanoat (7.07 g, 74% Reinheit, 16.0 mmol, Beispiel 309A) in ie/f-Butanol (97 ml) wurde mit Raney-Nickel (937 mg, 16.0 mmol) versetzt und drei Tage bei Normaldruck hydriert. Anschließend wurde der Katalysator über Kieselgur abfiltriert und zweimal mit ie f-Butanol (jeweils 30 ml) nachgewaschen. Das Filtrat wurde eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wur- den 6.21 g (65% Reinheit, 76% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.16 min; MS (ESIpos): m/z = 334 [M+H]+ Beispiel 311A
(+/-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-[2-(difluormethoxy)- 6-fluorphenyl]pentanoat (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (500 mg, 1.46 mmol, darstellbar gemäß WO 2016/146602, Beispiel 3A) in DMF (6 ml) wurden HATU (639 mg, 1.68 mmol) und DIPEA (570 μΙ, 3.3 mmol) gegeben und das Gemisch wurde 15 min bei RT gerührt. Anschließend wurde (+/-)-ie/f-Butyl-5-amino-4-[2-(difluormethoxy)-6-fluorphenyl]pentanoat (751 mg, 65% Reinheit, 1.46 mmol, Beispiel 310A), gelöst in DMF (3 ml) hinzugegeben, und das Gemisch wurde 16 h bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Ethylacetat
und Wasser (jeweils 100 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat (80 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter Natriumchlorid-Lösung (100 ml) gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash- Säulenchromatographie (50 g Kieselgel Biotage Snap-Cartridge Ultra, Cyclohexan/Ethylacetat- Gradient 97:3— > 7:3, Isolera One) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 749 mg (88% Reinheit, 69% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.54 min; MS (ESIpos): m/z = 657/659 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.92 (t, 1 H), 7.94 (d, 1 H), 7.86 (dd, 1 H), 7.70 (br. s, 1 H), 7.58-7.45 (m, 5H), 7.45-7.22 (m, 2H), 7.16-6.99 (m, 2H), 3.92-3.67 (m, 2H), 3.60-3.44 (m, 1 H), 2.22-1.87 (m, 7H), 1.35 (s, 9H).
Trennung der Enantiomere:
Die Titelverbindung (640 mg, 88% Reinheit) wurde in einem warmen Gemisch aus Acetonitril (7 ml) und Isopropanol (3 ml) gelöst und mittels präparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 312A und 313A) [Säule: Daicel Chiralpak IE, 5 μηη, 250 mm x 20 mm; Fluss: 15 ml/min; Detektion: 210 nm; Temperatur: 25 °C; Injektion: 0.50 ml; Eluent: 70% Heptan; 30% Isopropanol; Laufzeit 19 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde in Aceton itril/Wasser lyophilisiert. Beispiel 312A
(+)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-[2-(difluormethoxy)-6- fluorphenyljpentanoat (Enantiomer 1)
Bei der in Beispiel 31 1A beschriebenen Enantiomerentrennung wurden drei Chargen der Titelverbindung als früher eluierendes Enantiomer erhalten: 78 mg (Charge 1 , 76% Reinheit, ee-Wert 99%, siehe Drehwert und LC-MS), 21 mg (Charge 2, 75% Reinheit, ee-Wert 92%) und 186 mg (Charge 3, 88% Reinheit, ee-Wert 94%).
[a]D 20 = +24.6°, 589 nm, c = 0.33 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.56 min; MS (ESIpos): m/z = 657/659 [M+H]+
Beispiel 313A
(-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-[2-(difluormethoxy)-6- fluorphenyljpentanoat (Enantiomer 2)
Bei der in Beispiel 31 1A beschriebenen Enantiomerentrennung wurden zwei Chargen der Titelverbindung als später eluierendes Enantiomer erhalten: 76 mg (Charge 1 , 97% Reinheit, ee-Wert 95%) und 1 18 mg (Charge 2, 97% Reinheit, ee-Wert 99%, siehe Drehwert und LC-MS)
[a]D 20 = -29.1 °, 589 nm, c = 0.34 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.56 min; MS (ESIpos): m/z = 657/659 [M+H]+
Beispiel 314A
(+/-)-ie/f-Butyl-4-cyan-4-(2,5-difluorphenyl)butanoat (Racemat)
Unter Argonatmosphäre wurde zur Herstellung einer LDA-Lösung Diisopropylamin (10 ml, 74 mmol) in THF (44 ml) bei -78 °C vorgelegt und langsam mit einer n-Butyllithium Lösung (1.6 M in Hexan, 44 ml, 70 mmol) versetzt. Nach erfolgter Zugabe wurde die Lösung 10 min bei 0 °C nachgerührt. Diese LDA-Lösung wurde langsam zu einer auf -78 °C gekühlten Lösung von (2,5- Difluorphenyl)acetonitril (8.1 ml, 98% Reinheit, 64 mmol) in THF (120 ml) getropft. Nach vollständiger Zugabe wurde das Kältebad entfernt, das Reaktionsgemisch auf 0 °C kommen gelassen und nach 15 min wieder auf -78 °C gekühlt. Dann wurde langsam eine Lösung von ierf-Butyl-3- brompropanoat (13 ml, 97% Reinheit, 77 mmol) in 44 ml THF zugetropft und das Gemisch 1 h bei -78 °C nachgerührt, dann auf RT erwärmen gelassen und über Nacht bei RT nachgerührt. Zur Aufarbeitung wurde Ammoniumchlorid-Lösung (300 ml, 10% in Wasser) hinzugefügt. Das Gemisch wurde 5 Minuten kräftig verrührt und dann zweimal mit Ethylacetat extrahiert. Die vereinig- ten organischen Phasen wurden nacheinander mit je zweimal 1 M Salzsäure, einer gesättigten Natriumbicarbonat-Lösung und einer gesättigten Natriumchlorid-Lösung gewaschen. Die organische Phase wurde dann über Natriumsulfat getrocknet und am Rotationsverdampfer eingeengt. Der Rückstand wurde in wenig DMSO gelöst und in 4 Portionen mittels präparativer HPLC (Methode 30) gereinigt. Die produkthaltigen Fraktionen wurden vereint und am Rotationverdampfer eingeengt. Es wurden 8.05 g (100% Reinheit, 45% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.1 1 min; MS (ESIpos): m/z = 282 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.381 (16.00), 2.313 (0.86), 2.332 (1.28), 2.350 (0.44), 4.414 (0.63), 7.353 (0.40), 7.366 (0.62), 7.374 (0.51 ), 7.377 (0.49).
Beispiel 315A
(+/-)-fe/f-Butyl-5-amino-4-(2,5-difluorphenyl)pentanoat (Racemat)
(+/-)-ierf-Butyl-4-cyan-4-(2,5-difluorphenyl)butanoat {Racemat, 8.05 g, 100% Reinheit, 28.6 mmol, Beispiel 314A) wurde in ierf-Butanol (210 ml, 2.2 mol) und Methanol (9.3 ml) vorgelegt und mit Raney-Nickel (1.68 g, 28.6 mmol) versetzt. Das Reaktionsgemisch wurde 9 Tage unter Normaldruck Wasserstoff gerührt. Trotz unvollständiger Umsetzung wurde die Reaktion abgebrochen. Der Katalysator wurde über Kieselgur abfiltriert und dreimal mit Methanol (jeweils 30 ml) gewaschen. Das Filtrat wurde einrotiert und der Rückstand in Ethylacetat (200 ml) aufgenommen. Die organische Phase wurde zweimal mit 1 M Salzsäure (jeweils 200 ml) extrahiert. Die vereinigten wässrigen Phasen wurden durch langsame Zugabe von Natriumbicarbonat auf pH 8 gebracht und zweimal mit Dichlormethan (jeweils 200 ml) extrahiert. Die vereinten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt. Es wurden 3.84 g (100% Reinheit, 47% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.12 min; MS (ESIpos): m/z = 286 [M+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 7.29-7.04 (m, 3H), 2.92-2.84 (m, 1 H), 2.79-2.67 (m, 2H), 2.05-1.91 (m, 3H), 1.74-1.61 (m, 1 H), 1.35 (s, 9H).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.1 1 1 (0.42), 1.354 (16.00), 2.012 (1.26), 2.025 (0.85), 2.728 (0.47), 2.736 (0.51 ), 2.747 (0.69), 2.751 (0.69).
Beispiel 316A
(+/-)-ie f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]am
phenyl)pentanoat (Racemat)
Zu einer Lösung von 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (1.01 g, 2.94 mmol, darstellbar gemäß WO 2016/146602, Beispiel 3A) in DMF (34 ml) wurden HATU (1.34 g, 3.53 mmol) und DIPEA (1.2 ml, 7.1 mmol) hinzugegeben. Das Gemisch wurde 15 min bei RT gerührt. Eine Lösung von (+/-)-ie/f-Butyl-5-amino-4-(2,5-difluorphenyl)pentanoat (840 mg, 2.94 mmol, Beispiel 315A) in wenig DMF wurde hinzugefügt und die Reaktionsmischung 2 h bei 60 °C gerührt, dann auf RT abkühlen gelassen. Das Gemisch wurde mit Ethylacetat verdünnt und nacheinander zweimal mit 1 M-Salzsäure und zweimal mit einer gesättigten Natriumhydrogencarbonat-Lösung gewaschen. Die organische Phase wurde über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in wenig DMSO gelöst und mittels präparativer HPLC (Methode 30) gereinigt. Die geeigneten Fraktionen wurden zusammen eingeengt und im Vakuum getrocknet. Es wurde 1.16 g (100% Reinheit, 65% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.52 min; MS (ESIpos): m/z = 609/61 1 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.88 (t,1 H), 7.94 (d, 1 H); 7.85 (dd, 1 H), 7.65 (br. s, 1 H), 7.55-7.46 (m, 5H), 7.32 (ddd, 1 H), 7.23 (dt, 1 H), 7.17-7.07 (m, 1 H), 3.81 -3.63 (m, 2H), 3.43-3.33 (m, 1 H), 2.20-1.96 (m. 6H), 1.86-1.74 (m, 1 H), 1.37 (m, 9H).
Trennung der Enantiomere:
Die Titelverbindung (1.09 g) wurde in einem Gemisch (12 ml) aus Acetonitril, Methanol und Dich- lormethan gelöst und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 317A und 318A) [Säule: Daicel Chiralcel OX-H, 5 μηι, 250 mm x 30 mm; Fluss: 100 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 0.30 ml; Eluent: 81 % Kohlendioxid; 19% Methanol; Laufzeit 9.5 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde aus Acetonitril/Wasser lyophilisiert.
Beispiel 317A
ie f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2,5-^
pentanoat (Enantiomer 1)
Bei der unter Beispiel 316A beschriebenen Enantiomerentrennung wurden 432 mg (>99% Rein- heit, ee-Wert >99%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
LC-MS (Methode 1 ): Rt = 2.51 min; MS (ESIpos): m/z = 609/61 1 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.20), 0.008 (0.20), 1.030 (0.22), 1.045 (0.22), 1.369 (16.00), 1.806 (0.17), 2.012 (0.19), 2.029 (0.20), 2.049 (0.19), 2.061 (0.17), 2.087 (0.74), 2.104 (1.03), 2.123 (0.70), 3.368 (0.17), 3.673 (0.18), 3.687 (0.27), 7.1 17 (0.18), 7.200 (0.20), 7.21 1 (0.21 ), 7.223 (0.32), 7.234 (0.32), 7.293 (0.17), 7.300 (0.20), 7.306 (0.21 ), 7.315 (0.30), 7.324 (0.21 ), 7.330 (0.20), 7.338 (0.17), 7.489 (0.21 ), 7.501 (0.68), 7.508 (0.70), 7.519 (2.70), 7.529 (0.88), 7.533 (0.82), 7.833 (0.36), 7.838 (0.33), 7.856 (0.56), 7.861 (0.54), 7.929 (1.06), 7.952 (0.68), 8.860 (0.22), 8.874 (0.39), 8.888 (0.21 ).
Beispiel 318A
ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2,5-difluorphenyl)- pentanoat {Enantiomer 2)
Bei der unter Beispiel 316A beschriebenen Enantiomerentrennung wurden 366 mg (97% Reinheit, ee-Wert 94%) der Titelverbindung als später eluierendes Enantiomer erhalten.
LC-MS (Methode 1 ): Rt = 2.51 min; MS (ESIpos): m/z = 609/61 1 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.030 (0.69), 1.046 (0.70), 1.369 (16.00), 1.806 (0.20), 1.814 (0.17), 1.831 (0.17), 1.999 (0.17), 2.012 (0.22), 2.029 (0.23), 2.049 (0.21 ), 2.061 (0.19), 2.088 (0.84), 2.104 (1.17), 2.124 (0.83), 3.369 (0.20), 3.674 (0.22), 3.688 (0.31 ), 3.701 (0.18), 3.737 (0.19), 3.752 (0.18), 7.117 (0.22), 7.200 (0.21 ), 7.21 1 (0.23), 7.223 (0.35), 7.235 (0.34), 7.246 (0.17), 7.293 (0.19), 7.301 (0.23), 7.307 (0.24), 7.315 (0.34), 7.324 (0.24), 7.330 (0.23), 7.338 (0.18), 7.490 (0.25), 7.501 (0.74), 7.508 (0.77), 7.519 (3.02), 7.833 (0.37), 7.838 (0.35), 7.856 (0.57), 7.861 (0.57), 7.929 (1.08), 7.952 (0.68), 8.860 (0.25), 8.875 (0.45), 8.888 (0.24).
Ausführungsbeispiele:
Beispiel 1
(+/-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-phenylpropansäure (Racemat)
Zu einer Lösung aus (+/-)-Methyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- hydratropaat (153 mg, 304 μηηοΙ, Beispiel 153A) in THF (3.8 ml) und Methanol (750 μΙ) wurde bei RT 1 M Natronlauge (1.8 ml, 1.8 mmol) gegeben, und das Gemisch wurde 2 h bei RT gerührt. Anschließend wurde das Gemisch mit TFA (160 μΙ, 2.1 mmol) auf pH 3 eingestellt und mittels praparativer HPLC (Methode 15) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und lyophilisiert. Es wurden 147 mg (100% Reinheit, 99% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.84 min; MS (ESIpos): m/z = 489/491 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.54), 0.008 (1.39), 2.146 (3.32), 3.839 (1.33), 3.859 (2.67), 3.875 (2.66), 3.890 (1.71 ), 3.987 (2.43), 4.006 (3.60), 4.025 (1.43), 7.300 (0.95), 7.310 (1.56), 7.322 (1.64), 7.332 (1.06), 7.359 (0.74), 7.383 (13.34), 7.393 (16.00), 7.493 (1.15), 7.497 (1.29), 7.502 (1.24), 7.506 (4.09), 7.514 (3.84), 7.526 (1 1.82), 7.530 (1 1.18), 7.534 (7.60), 7.538 (6.08), 7.542 (5.18), 7.550 (1.32), 7.844 (2.39), 7.849 (2.09), 7.866 (3.62), 7.871 (3.43), 7.938 (6.67), 7.961 (4.21 ), 8.953 (1.27), 8.967 (2.44), 8.981 (1.28).
Trennung der Enantiomere:
Die Titelverbindung (120 mg) wurde in Methanol (20 ml) gelöst und mittels praparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 2 und 3) [Säule: Daicel Chiralcel OJ, 10 μηη, 250 mm x 20 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 1.5 ml; Eluent: 70% Kohlendioxid / 30% Methanol; Laufzeit 7 min, isokratisch]. Die vereinigten Ziel- fraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet.
Beispiel 2
(-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-phenylpropansäure (Enantio- mer 1)
Bei der in Beispiel 1 beschriebenen Enantiomerentrennung wurden 48 mg (99% Reinheit, ee-Wert 100%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -52.4°, 589 nm, c = 0.37 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.87 min; MS (ESIpos): m/z = 489/491 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.61 ), 0.008 (1.51 ), 2.145 (3.84), 3.390 (2.69), 3.838 (1.45), 3.858 (2.86), 3.873 (2.92), 3.888 (1.79), 3.986 (2.43), 4.005 (3.66), 4.025 (1.42), 5.754 (0.57), 7.300 (1.16), 7.310 (1.79), 7.321 (1.90), 7.331 (1.21 ), 7.359 (0.97), 7.382 (14.91 ), 7.392 (16.00), 7.490 (1.45), 7.494 (1.60), 7.503 (4.56), 7.51 1 (4.16), 7.523 (12.91 ), 7.527 (12.16), 7.532 (8.26), 7.536 (6.68), 7.539 (5.57), 7.547 (1.36), 7.746 (0.83), 7.838 (2.39), 7.844 (2.10), 7.861 (3.53), 7.866 (3.29), 7.935 (6.47), 7.957 (4.09), 8.950 (1.42), 8.963 (2.65), 8.977 (1.37), 12.636 (0.94).
Beispiel 3
(+)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-phenylpropansäure (Enantio- mer 2)
Bei der in Beispiel 1 beschriebenen Enantiomerentrennung wurden 45 mg (98% Reinheit, ee-Wert 97%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = +53.3°, 589 nm, c = 0.36 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.86 min; MS (ESIpos): m/z = 489/491 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.72), 0.008 (2.08), 2.145 (4.80), 3.837 (1.65), 3.857 (3.14), 3.873 (3.24), 3.887 (1.86), 3.983 (2.1 1 ), 4.002 (3.05), 4.021 (1.23), 5.754 (1.78), 7.308 (2.12), 7.319 (2.33), 7.329 (1.55), 7.358 (1.70), 7.380 (15.29), 7.391 (16.00), 7.479 (1.30), 7.490 (2.31 ), 7.493 (2.50), 7.503 (5.97), 7.51 1 (5.63), 7.523 (15.28), 7.527 (14.25), 7.531 (9.97), 7.535 (8.05), 7.539 (6.55), 7.547 (1.78), 7.743 (1.09), 7.838 (2.93), 7.843 (2.58), 7.860 (4.19), 7.865 (3.87), 7.934 (7.38), 7.957 (4.61 ), 8.948 (1.54), 8.961 (2.62), 8.975 (1.38).
Beispiel 4
(+/-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(2-chlorphenyl)propansäure
(Racemat)
Zu einer Lösung aus (+/-)-Ethyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(2- chlorphenyl)propanoat (2.20 g, 3.99 mmol, Beispiel 154A) in THF (32 ml) und Methanol (6.0 ml) wurde bei RT 1 M Natronlauge (24 ml, 24 mmol) gegeben, und das Gemisch wurde 2 h bei RT gerührt. Anschließend wurde das Gemisch mit Wasser und Ammoniumchlorid-Lösung (jeweils 100 ml) versetzt und geschüttelt. Die wässrige Phase wurde einmal mit Ethylacetat (100 ml) extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 1.89 g (99% Reinheit, 90% d. Th.) der Titelverbindung (Rohprodukt) erhalten. Etwa 100 mg des Rohproduktes wurden mittels präparativer HPLC (Methode 23) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden daraus 91 mg (100% Reinheit) der nachgereinigten Titelverbindung (siehe Analytik) erhalten.
LC-MS (Methode 1 ): Rt = 1.93 min; MS (ESIpos): m/z = 523/525 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.46), 0.008 (1.40), 1.194 (0.46), 1.21 1 (0.50), 2.164 (12.91 ), 3.881 (1.03), 3.897 (1.02), 3.984 (0.85), 3.998 (1.39), 4.013 (1.29), 4.032 (0.91 ), 4.483 (1.81 ), 4.501 (3.08), 4.520 (1.58), 5.754 (3.50), 7.319 (0.99), 7.338 (2.47), 7.353 (2.26), 7.370 (2.06), 7.388 (2.94), 7.405 (1.31 ), 7.479 (0.74), 7.492 (5.81 ), 7.495 (5.88), 7.504 (8.98), 7.51 1 (8.29), 7.514 (6.53), 7.523 (16.00), 7.528 (13.36), 7.536 (7.86), 7.547 (1.66), 7.748 (0.97), 7.838 (2.95), 7.844 (2.54), 7.860 (4.43), 7.866 (4.14), 7.934 (7.94), 7.956 (4.98), 8.959 (1.61 ), 8.973 (3.20), 8.987 (1.57), 12.794 (0.56).
Trennung der Enantiomere:
Die Titelverbindung (1.79 g, Rohprodukt) wurde in Methanol (80 ml) gelöst, filtriert und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 5 und 6) [Säule: Daicel Chiralcel OJ-H, 5 μηη, 250 mm x 30 mm; Fluss: 175 ml/min; Detektion: 210 nm; Temperatur: 38 °C; Injektion: 3 ml; Eluent: 67% Kohlendioxid / 33% Methanol; Laufzeit 4.5 min, isok- ratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet.
Beispiel 5
(-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(2-chlorphenyl)propansäure (Enantiomer 1)
Bei der in Beispiel 4 beschriebenen Enantiomerentrennung wurde die vorgereinigte Titelverbindung erhalten und mittels präparativer HPLC (Methode 24) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt und lyophilisiert. Es wurden 819 mg (100% Reinheit, ee-Wert 95%) der Titelverbindung erhalten.
[a]D 20 = -37.1 °, 589 nm, c = 0.39 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.91 min; MS (ESIpos): m/z = 523/525 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (1.48), 2.164 (12.99), 3.897 (1.37), 3.998 (2.04), 4.015 (1.92), 4.033 (1.57), 4.483 (2.1 1 ), 4.502 (3.31 ), 4.521 (1.70), 7.318 (1.23), 7.334 (2.69), 7.353 (2.42), 7.370 (2.04), 7.385 (2.99), 7.404 (1.34), 7.491 (6.69), 7.495 (6.78), 7.504 (9.81 ), 7.51 1 (8.77), 7.523 (16.00), 7.529 (12.90), 7.537 (7.20), 7.540 (5.52), 7.548 (1.34), 7.750 (1.01 ), 7.840 (2.91 ), 7.845 (2.34), 7.862 (4.23), 7.868 (3.62), 7.935 (7.28), 7.957 (4.47), 8.960 (1.77), 8.974 (3.16), 8.989 (1.52).
Beispiel 6
(+)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(2-chlorphenyl)propansäure (Enantiomer 2)
Bei der in Beispiel 4 beschriebenen Enantiomerentrennung wurde die vorgereinigte Titelverbindung erhalten und mittels präparativer HPLC (Methode 24) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt und lyophilisiert. Es wurden 638 mg (100% Reinheit, ee-Wert 100%) der Titelverbindung erhalten. [a]D 20 = +45.6°, 589 nm, c = 0.44 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.94 min; MS (ESIpos): m/z = 523/525 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.12), 0.008 (1.86), 2.164 (13.01 ), 3.882 (1.00), 3.899 (1.02), 3.983 (0.83), 3.997 (1.35), 4.014 (1.32), 4.033 (0.88), 4.483 (1.84), 4.501 (3.12), 4.520 (1.60), 7.315 (0.92), 7.319 (1.02), 7.334 (2.42), 7.338 (2.53), 7.353 (2.23), 7.357 (2.23), 7.366 (1.90), 7.370 (2.08), 7.385 (2.85), 7.389 (2.95), 7.404 (1.34), 7.478 (0.74), 7.492 (5.61 ), 7.495 (5.61 ), 7.504 (8.86), 7.51 1 (8.04), 7.514 (6.19), 7.523 (16.00), 7.527 (13.18), 7.531 (9.63), 7.536 (7.61 ), 7.539 (6.12), 7.547 (1.54), 7.748 (0.85), 7.838 (2.88), 7.843 (2.47), 7.860 (4.34), 7.865 (4.05), 7.934 (7.79), 7.956 (4.95), 8.958 (1.67), 8.973 (3.28), 8.987 (1.62), 12.797 (2.25).
Beispiel 7
(+/-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(2-methoxyphenyl)propan- säure (Racemat)
Zu einer Lösung aus (+/-)-Methyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2- (2-methoxyphenyl)propanoat (1.1 1 g, 2.08 mmol, Beispiel 155A) in THF (18 ml) und Methanol (3.6 ml) wurde bei RT 1 M Natronlauge (12 ml, 12 mmol) gegeben, und das Gemisch wurde 3 h bei RT gerührt. Anschließend wurde das Gemisch mit Wasser (100 ml) und 1 M Salzsäure (15 ml) versetzt und geschüttelt. Die wässrige Phase wurde einmal mit Ethylacetat (100 ml) extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 1.08 g (100% Reinheit, 100% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.85 min; MS (ESIpos): m/z = 519/521 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.61 ), 1.181 (0.40), 1.908 (0.42), 2.176 (6.04), 3.162 (0.99), 3.175 (1.01 ), 3.735 (0.58), 3.750 (0.98), 3.769 (0.95), 3.784 (1.09), 3.797 (16.00), 3.91 1 (0.54), 3.925 (0.72), 3.929 (0.71 ), 3.944 (0.99), 3.958 (0.52), 3.962 (0.55), 3.976 (0.48), 4.267 (1.1 1 ), 4.286 (2.15), 4.305 (0.88), 6.937 (0.89), 6.955 (1.87), 6.974 (1.09), 7.019 (1.64), 7.039 (1.89), 7.260 (1.06), 7.279 (4.03), 7.298 (2.96), 7.478 (0.47), 7.490 (0.95), 7.504 (2.64), 7.51 1 (2.38), 7.524 (5.63), 7.530 (5.48), 7.537 (3.68), 7.550 (0.79), 7.774 (0.66), 7.837 (1.35), 7.843 (1.1 1 ), 7.860 (2.00), 7.865 (1.77), 7.932 (3.50), 7.955 (2.22), 8.845 (0.81 ), 8.860 (1.54), 8.873 (0.73), 12.396 (0.79).
Trennung der Enantiomere:
Die Titelverbindung (1.0 g) wurde in 45 ml eines Gemisches aus Methanol und Acetonitril gelöst und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 8 und 9) [Säule: Daicel Chiralcel OJ-H, 5 μηι, 250 mm x 20 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 0.4 ml; Eluent: 70% Kohlendioxid / 30% Methanol; Laufzeit 4 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet.
Beispiel 8
(-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(2-methoxy
(Enantiomer 1)
Bei der in Beispiel 7 beschriebenen Enantiomerentrennung wurden 450 mg (100 % Reinheit, ee-Wert 100%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -57.5°, 589 nm, c = 0.35 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.84 min; MS (ESIpos): m/z = 519/521 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.89), 2.073 (0.62), 2.176 (6.14), 3.750 (1.00), 3.768 (1.06), 3.784 (1.16), 3.797 (16.00), 3.943 (0.98), 4.267 (1.10), 4.286 (2.10), 4.305 (0.86), 6.935 (0.87), 6.938 (0.93), 6.954 (1.81 ), 6.957 (1.81 ), 6.973 (1.03), 6.975 (1.06), 7.020 (1 .67), 7.039 (1.87), 7.256 (0.87), 7.260 (1.12), 7.279 (4.03), 7.298 (2.95), 7.490 (1.05), 7.493 (1.02), 7.496 (1.01 ), 7.505 (2.81 ), 7.512 (2.56), 7.516 (1.71 ), 7.525 (5.79), 7.530 (5.58), 7.534 (4.52), 7.538 (3.81 ), 7.542 (2.84), 7.550 (0.83), 7.838 (1.39), 7.843 (1.16), 7.860 (2.02), 7.865 (1.80), 7.933 (3.43), 7.955 (2.18), 8.846 (0.84), 8.860 (1.54). Beispiel 9
(+)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(2-methoxyphenyl)propan- säure (Enantiomer 2)
Bei der in Beispiel 7 beschriebenen Enantiomerentrennung wurden 318 mg (99 % Reinheit, ee-Wert 100%) der Titelverbindung als später eluierendes Enantiomer erhalten. [a]D 20 = +60.3°, 589 nm, c = 0.36 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.84 min; MS (ESIpos): m/z = 519/521 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.62), 0.008 (0.54), 2.073 (1.05), 2.176 (6.09), 2.523 (0.51 ), 3.735 (0.50), 3.750 (0.84), 3.767 (0.73), 3.784 (0.75), 3.797 (16.00), 3.91 1 (0.48), 3.925 (0.66), 3.929 (0.67), 3.943 (0.92), 3.962 (0.53), 3.976 (0.46), 4.266 (1.00), 4.285 (2.02), 4.304 (0.83), 6.937 (0.86), 6.956 (1.82), 6.975 (1.04), 7.019 (1.64), 7.039 (1.88), 7.260 (0.98), 7.279 (3.91 ), 7.298 (2.92), 7.490 (0.78), 7.505 (2.48), 7.51 1 (2.19), 7.525 (5.43), 7.531 (5.42), 7.537 (3.79), 7.550 (0.84), 7.773 (0.66), 7.838 (1.35), 7.843 (1.13), 7.860 (2.02), 7.865 (1.87), 7.933 (3.53), 7.955 (2.27), 8.847 (0.74), 8.861 (1.48), 8.875 (0.72), 12.407 (0.66).
Beispiel 10
(+/-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(4-fluorphenyl)propansäure (Racemat)
Zu einer Lösung aus (+/-)-Methyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(4- fluorphenyl)propanoat (192 mg, 368 mol, Beispiel 156A) in THF (3.1 ml) und Methanol (630 μΙ) wurde bei RT 1 M Natronlauge (2.2 ml, 2.2 mmol) gegeben, und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch mit TFA (198 μΙ, 2.6 mmol) auf pH 3 eingestellt und eingeengt. Der Rückstand wurde in DMSO aufgenommen und mittels präparativer HPLC (Methode 15) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 143 mg (100% Reinheit, 76% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.88 min; MS (ESIpos): m/z = 507/509 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 2.086 (1.66), 2.158 (5.56), 3.802 (1.49), 3.854 (3.59), 3.871 (5.34), 3.885 (3.88), 4.000 (2.74), 4.019 (4.23), 4.039 (1.71 ), 5.754 (1.63), 7.190 (4.07), 7.212 (8.57), 7.234 (4.78), 7.417 (5.22), 7.430 (6.34), 7.437 (5.67), 7.451 (4.32), 7.482 (0.91 ), 7.493 (2.23), 7.507 (6.82), 7.514 (5.93), 7.526 (16.00), 7.533 (15.50), 7.682 (0.75), 7.838 (2.91 ), 7.843 (2.58), 7.861 (4.53), 7.865 (4.06), 7.936 (7.1 1 ), 7.958 (4.55), 8.939 (2.18), 8.953 (4.25), 8.966 (2.12).
Trennung der Enantiomere:
Die Titelverbindung (120 mg) wurde in 12 ml eines Gemisches aus Acetonitril und Methanol gelöst und mittels präparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 1 1 und 12) [Säule: Daicel Chiralpak IA, 5 μηη, 250 mm x 4.6 mm; Fluss: 20 ml/min; Detektion: 220 nm; Temperatur: 23 °C; Injektion: 0.08 ml; Eluent: 55% Heptan / (45% Ethanol + 0.2% TFA + 1 % Wasser); isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und lyophilisiert.
Beispiel 11
3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(4-fluorphenyl)propansäure (Enan- tiomer 1 )
Bei der in Beispiel 10 beschriebenen Enantiomerentrennung wurde die vorgereinigte Titelverbindung als früher eluierendes Enantiomer (ee-Wert 100%) erhalten, anschließend in Acetonitril aufgenommen und mittels präparativer HPLC (Methode 15) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt und lyophilisiert. Es wurden 17 mg (100% Reinheit) der nachgerei- nigten Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.86 min; MS (ESIpos): m/z = 507/509 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.75), 0.008 (0.71 ), 2.157 (5.20), 2.327 (0.46), 2.366 (0.45), 2.523 (2.18), 2.670 (0.40), 3.800 (0.54), 3.853 (3.1 1 ), 3.870 (4.71 ), 3.884 (3.33), 3.998 (2.78), 4.017 (4.32), 4.037 (1.65), 7.189 (4.64), 7.21 1 (9.47), 7.233 (5.23), 7.407 (1.61 ), 7.415 (6.21 ), 7.421 (3.75), 7.429 (7.03), 7.437 (6.30), 7.445 (3.06), 7.451 (5.25), 7.480 (1.60), 7.491 (2.83), 7.496 (2.99), 7.506 (7.45), 7.513 (6.73), 7.525 (16.00), 7.530 (15.37), 7.534 (12.62), 7.538 (9.71 ), 7.542 (7.43), 7.550 (2.18), 7.682 (0.72), 7.836 (3.05), 7.842 (2.87), 7.859 (4.50), 7.864 (4.25), 7.934 (8.30), 7.956 (5.24), 8.937 (2.32), 8.951 (4.34), 8.965 (2.05), 12.695 (0.72). Beispiel 12
3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(4-fluorphenyl)propansäure (Enantiomer 2)
Bei der in Beispiel 10 beschriebenen Enantiomerentrennung wurde die vorgereinigte Titelverbindung als später eluierendes Enantiomer (ee-Wert 97%) erhalten, anschließend in Acetonitril auf- genommen und mittels präparativer HPLC (Methode 15) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt und lyophilisiert. Es wurden 18 mg (100% Reinheit) der nachgereinigten Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.86 min; MS (ESIpos): m/z = 507/509 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (0.59), 2.157 (4.91 ), 2.523 (1.21 ), 3.852 (2.68), 3.870 (4.44), 3.885 (3.20), 3.999 (2.66), 4.018 (4.37), 4.037 (1.66), 7.189 (4.59), 7.194 (2.01 ), 7.21 1 (9.94), 7.216 (2.80), 7.228 (2.29), 7.233 (5.54), 7.241 (0.79), 7.408 (0.93), 7.415 (5.86), 7.421 (3.02), 7.429 (6.70), 7.437 (5.97), 7.446 (2.54), 7.451 (5.00), 7.459 (0.67), 7.480 (0.87), 7.491 (2.04), 7.506 (6.75), 7.513 (5.85), 7.525 (16.00), 7.531 (15.10), 7.534 (12.59), 7.538 (9.63), 7.542 (7.55), 7.550 (2.09), 7.683 (0.64), 7.836 (3.1 1 ), 7.842 (2.87), 7.859 (4.78), 7.864 (4.59), 7.934 (9.03), 7.957 (5.79), 8.937 (2.16), 8.951 (4.37), 8.965 (2.06), 12.698 (1.19).
Beispiel 13
(+/-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-methyl-2-phenylpropansäure (Racemat)
Zu einer Lösung aus (+/-)-Ethyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2- methylhydratropaat (204 mg, 383 mol, Beispiel 157A) in THF (3.2 ml) und Methanol (1.6 ml) wurde bei RT 1 M Natronlauge (2.3 ml, 2.3 mmol) gegeben, und das Gemisch wurde 20 h bei RT gerührt. Anschließend wurde das Gemisch mit TFA (210 μΙ, 2.7 mmol) auf pH 3 eingestellt und mittels präparativer HPLC (Methode 20) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und lyophilisiert. Es wurden 195 mg (100% Reinheit,„100% d. Th.", lösungsmittelhal- tig) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.03 min; MS (ESIpos): m/z = 503/505 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.49), 0.008 (0.58), 1.647 (16.00), 2.072 (0.68), 2.1 16 (2.00), 3.806 (1.36), 3.818 (1.54), 3.840 (1.77), 3.852 (1.63), 4.176 (1.56), 4.194 (1.70), 4.210 (1.46), 4.228 (1.38), 5.754 (1.35), 7.274 (0.78), 7.292 (2.20), 7.310 (1.67), 7.364 (2.52), 7.384 (5.10), 7.402 (3.22), 7.434 (6.71 ), 7.452 (3.72), 7.477 (0.50), 7.489 (1.51 ), 7.504 (4.49), 7.510 (3.58), 7.522 (7.48), 7.535 (8.74), 7.552 (1.68), 7.682 (0.67), 7.832 (2.05), 7.837 (1.89), 7.854 (3.10), 7.860 (3.04), 7.931 (5.79), 7.953 (3.72), 8.650 (1.26), 8.663 (1.77), 8.667 (1.79), 8.680 (1.25).
Trennung der Enantiomere:
Die Titelverbindung (175 mg) wurde in Methanol (15 ml) gelöst und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 14 und 15) [Säule: Daicel Chiralpak AD-H, 5 μη-ι, 250 mm x 20 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 2.0 ml; Eluent: 70% Kohlendioxid / 30% Methanol; Laufzeit 10 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet.
Beispiel 14
(-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-methyl-2-phenylpropansäure (Enantiomer 1)
Bei der in Beispiel 13 beschriebenen Enantiomerentrennung wurden 71 mg (95% Reinheit, ee-Wert 100%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -42.9°, 589 nm, c = 0.40 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.05 min; MS (ESIpos): m/z = 503/505 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.44), 0.008 (0.44), 1.645 (16.00), 2.1 16 (2.04), 2.523 (0.51 ), 3.805 (1.39), 3.817 (1.53), 3.838 (1.78), 3.851 (1.61 ), 4.174 (1.57), 4.192 (1.69), 4.208 (1.45), 4.226 (1.35), 7.274 (0.81 ), 7.292 (2.21 ), 7.310 (1.68), 7.324 (0.46), 7.363 (2.56), 7.383 (5.10), 7.402 (3.25), 7.433 (6.81 ), 7.451 (3.69), 7.475 (0.57), 7.487 (1.54), 7.502 (4.49), 7.508 (3.60), 7.520 (7.86), 7.533 (8.79), 7.550 (1.62), 7.678 (0.63), 7.828 (1.94), 7.833 (1.79), 7.850 (2.98), 7.856 (2.90), 7.929 (5.51 ), 7.951 (3.67), 8.648 (1.26), 8.665 (1.77), 8.678 (1.22), 12.658 (1.38).
Beispiel 15
(+)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-methyl-2-phenylpropansäure (Enantiomer 2)
Bei der in Beispiel 13 beschriebenen Enantiomerentrennung wurden 69 mg (98% Reinheit, ee-Wert 100%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = +42.4°, 589 nm, c = 0.37 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.05 min; MS (ESIpos): m/z = 503/505 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.640 (16.00), 2.1 14 (2.43), 3.804 (1.41 ), 3.816 (1.55), 3.838 (1.81 ), 3.850 (1.64), 4.167 (1.52), 4.184 (1.64), 4.200 (1.40), 4.218 (1.31 ), 7.272 (0.86),
7.290 (2.36), 7.307 (1.81 ), 7.324 (0.42), 7.361 (2.70), 7.381 (5.38), 7.399 (3.40), 7.431 (7.1 1 ),
7.450 (3.85), 7.474 (0.63), 7.487 (1.65), 7.502 (4.79), 7.507 (3.87), 7.520 (8.30), 7.532 (9.51 ),
7.549 (1.86), 7.678 (0.78), 7.828 (2.05), 7.833 (1.89), 7.850 (3.15), 7.855 (3.08), 7.928 (5.67), 7.950 (3.76), 8.654 (1.26), 8.670 (1.87), 8.684 (1.24). Beispiel 16
(+/-)-2-({[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}methyl)-2-phenylbutansäure (Racemat)
Zu einer Lösung aus (+/-)-Methyl-2-({[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- methyl)-2-phenylbutanoat (540 mg, 1.02 mmol, Beispiel 158A) in THF (8.7 ml) und Methanol (1.7 ml) wurde bei RT 1 M Natronlauge (6.1 ml, 6.1 mmol) gegeben, und das Gemisch wurde 16 h bei RT gerührt. Anschließend wurde erneut Natronlauge (6.1 ml, 6.1 mmol) hinzugegeben, und das Gemisch wurde weitere 4 h bei RT gerührt. Anschließend wurde Methanol (5 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 40 °C gerührt. Anschließend wurde das Gemisch mit TFA auf pH 3 eingestellt und mittels praparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und lyophilisiert. Es wurden 495 mg (98% Reinheit, 92% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.07 min; MS (ESIpos): m/z = 517/519 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.883 (5.29), 0.901 (12.03), 0.919 (5.67), 2.085 (2.56), 2.101 (3.71 ), 2.1 18 (4.29), 2.138 (4.09), 2.158 (2.91 ), 2.175 (1.49), 2.193 (0.81 ), 2.689 (1.35), 2.731 (1.66), 2.890 (1.78), 3.947 (1.70), 3.960 (1.84), 3.981 (2.25), 3.994 (2.15), 4.206 (2.25), 4.223 (2.29), 4.240 (1.89), 4.257 (1.73), 7.274 (2.20), 7.291 (1.79), 7.349 (2.00), 7.370 (6.65), 7.387 (16.00), 7.404 (2.33), 7.475 (0.67), 7.487 (1.79), 7.501 (5.73), 7.507 (4.68), 7.519 (1 1.55), 7.529 (1 1.24), 7.546 (1.98), 7.642 (0.69), 7.827 (2.51 ), 7.832 (2.35), 7.849 (3.86), 7.854 (3.80), 7.923 (7.49), 7.946 (4.75), 8.501 (1.66), 8.516 (2.84), 8.530 (1.64).
Trennung der Enantiomere:
Die Titelverbindung (450 mg) wurde in Methanol (25 ml) gelöst und mittels praparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 17 und 18) [Säule: Daicel Chiralpak AD, 10 μιτι, 250 mm x 20 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 1.0 ml; Eluent: 78% Kohlendioxid / 22% Isopropanol; Laufzeit 10 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Beispiel 17
(-)-2-({[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}methyl)-2-phenylbutansäure (Enantiomer 1)
Bei der in Beispiel 16 beschriebenen Enantiomerentrennung wurden 156 mg (98% Reinheit, ee-Wert 100%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -10.0°, 436 nm, c = 0.32 g/100 ml, Chloroform
LC-MS (Methode 1 ): Rt = 2.02 min; MS (ESIpos): m/z = 517/519 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.882 (5.37), 0.901 (12.17), 0.919 (5.68), 2.084 (2.60), 2.100 (3.68), 2.1 18 (4.29), 2.137 (4.05), 2.157 (2.89), 2.174 (1.48), 2.193 (0.80), 2.327 (0.48), 2.669 (0.45), 2.890 (0.40), 3.781 (1.39), 3.946 (1.76), 3.958 (1.82), 3.980 (2.1 1 ), 3.993 (1.96), 4.205 (1.71 ), 4.222 (1.83), 4.238 (1.49), 4.256 (1.36), 7.273 (2.16), 7.290 (1.78), 7.349 (1.96), 7.369 (6.67), 7.386 (16.00), 7.403 (2.35), 7.473 (0.68), 7.486 (1.79), 7.500 (5.74), 7.507 (4.75), 7.518 (1 1.53), 7.528 (1 1.13), 7.636 (0.69), 7.826 (2.62), 7.831 (2.41 ), 7.848 (4.00), 7.853 (3.91 ), 7.923 (7.83), 7.945 (4.94), 8.499 (1.66), 8.515 (2.83), 8.530 (1.61 ).
Beispiel 18
(+)-2-({[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}methyl)-2-phenylbutansäure (Enantiomer 2)
Bei der in Beispiel 16 beschriebenen Enantiomerentrennung wurden 162 mg (98% Reinheit, ee-Wert 100%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = +1 1.4°, 436 nm, c = 0.35 g/100 ml, Chloroform
LC-MS (Methode 1 ): Rt = 2.02 min; MS (ESIpos): m/z = 517/519 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (3.29), 0.008 (3.13), 0.882 (5.46), 0.900 (12.52), 0.918 (5.74), 2.083 (2.56), 2.100 (3.63), 2.1 17 (4.23), 2.137 (4.00), 2.157 (2.86), 2.174 (1.47), 2.192 (0.78), 2.327 (0.67), 2.366 (0.54), 2.518 (2.78), 2.523 (2.15), 2.669 (0.64), 2.674 (0.48), 2.710 (0.52), 3.555 (5.62), 3.945 (1.41 ), 3.958 (1.52), 3.980 (1.88), 3.992 (1.71 ), 4.204 (1.66), 4.221 (1.78), 4.238 (1.44), 4.255 (1.31 ), 7.258 (0.94), 7.274 (2.1 1 ), 7.290 (1.78), 7.348 (2.01 ), 7.369 (6.76), 7.386 (16.00), 7.403 (2.20), 7.473 (0.73), 7.485 (1.95), 7.499 (5.70), 7.506 (5.02),
7.519 (1 1.25), 7.526 (10.60), 7.529 (10.35), 7.545 (1.82), 7.638 (0.61 ), 7.825 (2.75), 7.830 (2.53), 7.847 (4.20), 7.853 (4.05), 7.922 (7.81 ), 7.944 (4.98), 8.498 (1.69), 8.514 (2.76), 8.528 (1.61 ), 12.630 (0.48).
Beispiel 19
(+/-)-3-{[(6-Brom-3-methyl-2-phenyl^
säure (Racemat)
Zu einer Lösung aus (6-Brom-3-methyl-2-phenylchinolin-4-yl)(1 /-/-imidazol-1-yl)methanon (1.10 g, 2.80 mmol, darstellbar gemäß WO 2016/037954, Beispiel 2A, S. 58) in DMF (15 ml) wurde bei RT (+/-)-Methyl-3-amino-2-hydroxy-2-phenylpropanoat (602 mg, 3.08 mmol, darstellbar durch Zugabe von 1 M Natronlauge zu einem Gemisch aus (+/-)-Methyl-3-amino-2-hydroxy- 2-phenylpropanoat-Hydrochlorid (Beispiel 79A) in Wasser bis zu einem neutralen pH-Wert, gefolgt von Extraktion mit Dichlormethan und Einengen der organischen Phase) gegeben. Anschließend wurde langsam unter Rühren eine 1 M Lösung aus Kalium-ie f-butylat (7.0 ml, 7.0 mmol) in THF hinzugetropft, und das Reaktionsgemisch wurde 2 Tage bei RT gerührt. Anschließend wurde das Gemisch mit 10%-iger Zitronensäure (150 ml) versetzt und zweimal mit Ethyl- acetat (jeweils 100 ml) extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter wässriger Natriumchlorid-Lösung (100 ml) gewaschen und über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in einigen Millilitern eines Gemisches aus DMSO, Wasser und Acetonitril in der Wärme verrührt, und der vorhandene Feststoff wurde abfiltriert. Das Filtrat wurde mittels präparativer HPLC (Methode 22) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 149 mg (100% Reinheit, 1 1 % d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.75 min; MS (ESIpos): m/z = 505/507 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.87), 0.008 (1.97), 1.211 (0.43), 2.085 (2.34), 2.366 (0.84), 2.710 (0.84), 3.641 (0.45), 3.964 (4.57), 3.985 (3.50), 3.998 (3.54), 4.019 (3.77), 4.032 (3.54), 4.188 (2.47), 4.205 (2.62), 4.222 (2.06), 4.238 (1.91 ), 5.753 (6.52), 7.299 (0.99),
7.317 (2.55), 7.335 (2.34), 7.370 (4.53), 7.389 (6.61 ), 7.407 (2.90), 7.473 (0.88), 7.484 (1.84), 7.488 (1.95), 7.498 (5.68), 7.505 (5.64), 7.518 (16.00), 7.530 (8.94), 7.640 (9.05), 7.658 (8.26), 7.662 (6.09), 7.809 (3.05), 7.814 (2.81 ), 7.831 (4.48), 7.837 (4.40), 7.907 (9.09), 7.929 (5.81 ), 8.669 (1.65), 8.683 (2.81 ), 8.697 (1.59).
Trennung der Enantiomere:
Die Titelverbindung (140 mg) wurde in 17 ml eines Gemisches aus Methanol und Acetonitril gelöst und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 20 und 21 ) [Säule: Daicel Chiralpak AZ-H, 5 μηι, 250 mm x 20 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 0.5 ml; Eluent: 70% Kohlendioxid / 30% Methanol; Lauf- zeit 8 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet.
Beispiel 20
(+)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-hydroxy-2-phenylpropansäure (Enantiomer 1)
Bei der in Beispiel 19 beschriebenen Enantiomerentrennung wurde die vorgereinigte Titelverbindung als früher eluierendes Enantiomer (ee-Wert 98%) erhalten und mittels präparativer HPLC (Methode 23) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt und lyophilisiert. Es wurden 19 mg (95% Reinheit) der Titelverbindung erhalten.
[a]D 20 = +22.3°, 589 nm, c = 0.16 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 0.93 min; MS (ESIpos): m/z = 505/507 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 2.091 (2.15), 3.987 (1.42), 3.998 (1.50), 4.021 (1.95), 4.032 (1.77), 4.189 (2.04), 4.205 (2.06), 4.223 (1.62), 4.239 (1.51 ), 4.964 (0.51 ), 7.317 (2.50), 7.335 (2.35), 7.370 (4.32), 7.389 (6.47), 7.408 (2.86), 7.488 (1.95), 7.499 (5.60), 7.506 (5.19), 7.519 (16.00), 7.641 (8.95), 7.659 (7.59), 7.810 (2.80), 7.833 (4.18), 7.838 (4.16), 7.908 (8.05), 7.930 (5.10), 8.671 (1.74), 8.684 (2.84).
Beispiel 21
(-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-hydroxy-2-phenylpropansäure (Enantiomer 2)
Bei der in Beispiel 19 beschriebenen Enantiomerentrennung wurde die vorgereinigte Titelverbin- dung als später eluierendes Enantiomer (ee-Wert 96%) erhalten und mittels präparativer HPLC (Methode 23) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt und lyophilisiert. Es wurden 21 mg (96% Reinheit) der Titelverbindung erhalten.
[a]D 20 = -18.9°, 589 nm, c = 0.34 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 0.93 min; MS (ESIpos): m/z = 505/507 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 2.096 (2.18), 3.987 (1.37), 3.999 (1.50), 4.021 (1.98), 4.033 (1.83), 4.190 (1.98), 4.207 (2.14), 4.224 (1.65), 4.241 (1.49), 7.300 (1.10), 7.317 (2.65), 7.335 (2.43), 7.370 (4.60), 7.389 (6.63), 7.407 (3.06), 7.476 (0.94), 7.491 (2.08), 7.501 (5.89), 7.508 (5.53), 7.521 (16.00), 7.641 (8.97), 7.660 (7.96), 7.720 (0.57), 7.815 (2.82), 7.820 (2.60), 7.837 (4.27), 7.842 (4.07), 7.91 1 (7.89), 7.933 (5.02), 8.673 (1.77), 8.687 (3.01 ), 8.701 (1.71 ).
Beispiel 22
(+/-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-[3-(trifluormethyl)phenyl]- propansäure (Racemat)
Zu einer Lösung aus (+/-)-Ethyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-[3- (trifluormethyl)phenyl]propanoat (35 mg, 59.8 μηηοΙ, Beispiel 159A) in THF (510 μΙ) und Methanol (100 μΙ) wurde bei RT 1 M Natronlauge (360 μΙ, 360 μηηοΙ) gegeben, und das Gemisch wurde vier Tage bei RT gerührt. Anschließend wurde das Gemisch mit TFA (32 μΙ, 420 μηηοΙ) auf pH 3 eingestellt und danach eingeengt. Der Rückstand wurde in DMSO aufgenommen und mittels präparativer HPLC (Methode 15) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und lyophilisiert. Es wurden 147 mg (100% Reinheit, 99% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.07 min; MS (ESIneg): m/z = 557/559 [M-H]"
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 2.083 (1.10), 3.925 (1.15), 3.938 (1.69), 3.957 (2.04), 3.972 (1.05), 4.125 (1.30), 4.145 (2.27), 4.164 (0.90), 7.517 (16.00), 7.617 (0.69), 7.636 (1.82), 7.655 (1.80), 7.685 (2.15), 7.720 (6.05), 7.738 (1.73), 7.838 (1.37), 7.843 (1.35), 7.861 (2.08), 7.866 (2.10), 7.933 (3.89), 7.955 (2.44), 8.949 (0.93), 8.962 (1.72), 8.975 (0.94).
Beispiel 23
(+/-)-2-(1 ,3-Benzodioxol-5-yl)-3-{[(6-brom
säure (Racemat)
Zu einer Lösung aus (+/-)-Ethyl-2-(1 ,3-benzodioxol-5-yl)-3-{[(6-brom-3-methyl-2-phenylchinolin- 4-yl)carbonyl]amino}propanoat (1 10 mg, 196 μmol, Beispiel 160A) in THF (1.7 ml) und Methanol (330 μΙ) wurde bei RT 1 M Natronlauge (1.2 ml, 1.2 mmol) gegeben, und das Gemisch wurde vier Tage bei RT gerührt. Anschließend wurde das Gemisch mit TFA (1 10 μΙ, 1.4 mmol) auf pH 3 eingestellt und mittels präparativer HPLC (Methode 15) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und im Vakuum getrocknet. Es wurden 84 mg (98% Reinheit, 79% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 0.96 min; MS (ESIpos): m/z = 533/535 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.23), 0.008 (1.80), 2.185 (7.83), 2.328 (0.82), 2.367 (0.43), 2.523 (2.13), 2.670 (0.77), 3.823 (3.68), 3.906 (3.23), 3.925 (4.20), 3.944 (1.59), 4.869 (0.78), 5.966 (0.46), 6.000 (16.00), 6.830 (3.16), 6.835 (3.38), 6.851 (5.93), 6.855 (6.40), 6.893 (1 1.41 ), 6.913 (5.88), 6.935 (8.97), 6.938 (8.35), 7.483 (0.92), 7.495 (2.38), 7.510 (7.43), 7.516 (6.51 ), 7.529 (14.53), 7.536 (14.85), 7.540 (13.69), 7.556 (2.60), 7.725 (1.16), 7.841 (3.81 ), 7.846 (3.40), 7.864 (5.64), 7.869 (5.36), 7.938 (10.63), 7.961 (6.64), 8.928 (2.17), 8.941 (4.35), 8.955 (2.07).
Trennung der Enantiomere:
Die Titelverbindung (60 mg) wurde in Isopropanol (3 ml) gelöst und mittels präparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 24 und 25) [Säule: Chiralpak ID, 5 μηη, 250 mm x 20 mm; Fluss: 9 ml/min; Detektion: 220 nm; Temperatur: 50 °C; Injektion: 0.25 ml; Eluent: 35% Heptan / (65% Isopropanol + 0.2% Essigsäure); Laufzeit 15 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde in Acetonitril/Wasser lyophilisiert.
Beispiel 24
2-(1 ,3-Benzodioxol-5-yl)-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbon
(Enantiomer 1)
Bei der in Beispiel 23 beschriebenen Enantiomerentrennung wurde die vorgereinigte Titelverbin- dung als früher eluierendes Enantiomer (ee-Wert 99%) erhalten und mittels präparativer HPLC (Methode 20) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde in Aceton itril/Wasser lyophilisiert. Es wurden 20 mg (98% Reinheit) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 0.97 min; MS (ESIpos): m/z = 533/535 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: δ [ppm] = 8.94 (t, 1 H), 7.95 (d, 1 H), 7.86 (d, 1 H), 7.72 (br. s, 1 H), 7.60-7.46 (m, 5H), 6.98-6.81 (m, 3H), 6.00 (s, 2H), 3.98-3.76 (m, 3H), 2.19 (br. s, 3H).
Beispiel 25
2-(1 ,3-Benzodioxol-5-yl)-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}propansäure (Enantiomer 2)
Bei der in Beispiel 23 beschriebenen Enantiomerentrennung wurde die vorgereinigte Titelverbindung als später eluierendes Enantiomer (ee-Wert 94%) erhalten und mittels präparativer HPLC (Methode 20) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde in Aceton itril/Wasser lyophilisiert. Es wurden 22 mg (98% Reinheit) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 0.97 min; MS (ESIpos): m/z = 533/535 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.29), 0.008 (1.90), 2.185 (7.94), 2.231 (0.87), 2.328 (0.42), 2.523 (1.81 ), 2.670 (0.47), 3.906 (3.19), 3.925 (4.18), 3.944 (1.62), 5.966 (0.64), 6.000 (16.00), 6.831 (3.05), 6.834 (3.25), 6.851 (5.68), 6.855 (6.08), 6.893 (9.85), 6.913 (5.14), 6.935 (8.46), 6.938 (7.99), 7.484 (0.74), 7.495 (2.17), 7.510 (7.02), 7.517 (6.12), 7.529 (13.81 ), 7.537 (14.58), 7.540 (13.71 ), 7.556 (2.98), 7.725 (1.12), 7.842 (3.36), 7.847 (3.10), 7.864 (5.17), 7.869 (5.04), 7.939 (9.15), 7.961 (5.93), 8.928 (2.10), 8.942 (4.33), 8.955 (2.18).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.63 (br. s, 1 H), 8.94 (t, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.73 (br. s, 1 H), 7.58-7.44 (m, 5H), 6.98-6.80 (m, 3H), 6.00 (s, 2H), 3.99-3.75 (m, 3H), 2.19 (br. s, 3H).
Beispiel 26
(+/-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(3-chlorpyridin-2-yl)-2-methyl- propansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 2-(3-chlorpyridin-2-yl)-2-methylpropanoat (200 mg, 336 μηηοΙ, Beispiel 161A) in Dichlormethan (2.3 ml) wurde TFA (520 μΙ, 6.7 mmol) gegeben, und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyc- lohexan/Ethylacetat 8:2, Isolera One) vorgereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Der Rückstand wurde anschließend mittels prä- parativer HPLC (Methode 20) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der erhaltene Rückstand im Vakuum getrocknet. Es wurden 103 mg (98% Reinheit, 56% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.01 min; MS (ESIpos): m/z = 538/540 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (0.65), 1.195 (1.04), 1.21 1 (1.05), 1.261 (0.69), 1.277 (0.68), 1.683 (16.00), 2.190 (5.52), 3.930 (0.60), 3.950 (0.64), 4.471 (0.79), 4.488 (0.91 ), 4.505 (0.80), 4.523 (0.74), 7.373 (1.45), 7.384 (1.58), 7.392 (1.71 ), 7.404 (1.68), 7.493 (1.05), 7.508 (3.75), 7.514 (2.97), 7.526 (6.99), 7.538 (8.33), 7.555 (1.93), 7.845 (2.42), 7.849 (1.91 ), 7.867 (3.42), 7.872 (2.99), 7.899 (2.53), 7.902 (2.70), 7.919 (2.46), 7.922 (2.55), 7.930 (5.60), 7.953 (3.31 ), 8.527 (2.14), 8.538 (2.19), 8.696 (1.01 ), 8.712 (2.05), 8.728 (1.14).
Trennung der Enantiomere:
Die Titelverbindung (70 mg) wurde in Isopropanol (3 ml) gelöst und mittels präparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 27 und 28) [Säule: YMC Chiralart Amylose SA, 5 μηη, 250 mm x 30 mm; Fluss: 30 ml/min; Detektion: 220 nm; Temperatur: 40 °C; Injektion: 0.40 ml; Eluent: 50% Heptan / (50% Isopropanol + 0.2% Essigsäure); Laufzeit 15 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert.
Beispiel 27
3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(3-chlorpyridin
propansäure (Enantiomer 1)
Bei der in Beispiel 26 beschriebenen Enantiomerentrennung wurden 21 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
LC-MS (Methode 1 ): Rt = 1.89 min; MS (ESIpos): m/z = 538/540 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (0.93), 1.261 (0.44), 1.278 (0.45), 1.682 (16.00), 2.190 (4.78), 3.929 (0.64), 3.949 (0.69), 3.962 (0.61 ), 4.471 (0.86), 4.488 (0.94), 4.505 (0.85), 4.522 (0.76), 7.373 (1.63), 7.384 (1.74), 7.392 (1.79), 7.404 (1.73), 7.481 (0.56), 7.493 (1.46), 7.507 (4.12), 7.514 (3.34), 7.526 (7.32), 7.535 (7.73), 7.537 (7.88), 7.554 (1.42), 7.844 (2.38), 7.849 (1.82), 7.866 (3.25), 7.871 (2.70), 7.898 (2.33), 7.902 (2.45), 7.919 (2.31 ), 7.922 (2.43), 7.929 (4.94), 7.952 (2.91 ), 8.526 (2.14), 8.535 (2.04), 8.694 (1.14), 8.711 (2.1 1 ), 8.726 (1.06).
Beispiel 28
3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(3-chlorpyridin-2-yl)-2-methyl- propansäure (Enantiomer 2)
Bei der in Beispiel 26 beschriebenen Enantiomerentrennung wurden 23 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als später eluierendes Enantiomer erhalten.
LC-MS (Methode 2): Rt = 1.00 min; MS (ESIpos): m/z = 538/540 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (0.69), 1.682 (16.00), 2.185 (4.45), 2.189 (4.42), 3.928 (0.63), 3.949 (0.69), 4.470 (0.86), 4.488 (0.93), 4.505 (0.85), 4.522 (0.76), 7.373 (1.62), 7.384 (1.73), 7.392 (1.78), 7.404 (1.73), 7.480 (0.54), 7.493 (1.42), 7.507 (4.03), 7.514 (3.20),
7.525 (7.19), 7.535 (7.70), 7.554 (1.36), 7.844 (2.33), 7.849 (1.76), 7.866 (3.18), 7.871 (2.61 ), 7.899 (2.36), 7.902 (2.37), 7.919 (2.31 ), 7.922 (2.36), 7.930 (4.92), 7.952 (2.84), 8.523 (2.07),
8.526 (2.09), 8.535 (2.06), 8.694 (1.13), 8.71 1 (2.10), 8.727 (1.06). Beispiel 29
(+/-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(2-fluorphenyl)-2-methyl- propansäure (Racemat)
Zu einer Lösung aus (+/-)-Methyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2- (2-fluorphenyl)-2-methylpropanoat (189 mg, 354 μηιοΙ, Beispiel 162A) in THF (3.0 ml) und Methanol (1.5 ml) wurde bei RT 1 M Natronlauge (2.1 ml, 2.1 mmol) gegeben, und das Gemisch wurde 24 h bei RT gerührt. Anschließend wurde das Gemisch mit TFA (190 μΙ, 2.5 mmol) auf pH 3 eingestellt und mittels praparativer HPLC (Methode 20) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 154 mg (100% Reinheit, 83% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.01 min; MS (ESIpos): m/z = 521/523 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (1.16), 1.234 (0.44), 1.637 (16.00), 2.085 (1.65), 3.769 (1.31 ), 3.781 (1.45), 3.803 (1.60), 3.816 (1.47), 4.251 (1.65), 4.269 (1.82), 4.284 (1.61 ), 4.304 (1.50), 5.754 (2.61 ), 7.153 (1.37), 7.173 (1.89), 7.183 (1.90), 7.206 (3.41 ), 7.226 (1.71 ), 7.353 (1.43), 7.365 (1.34), 7.389 (2.06), 7.409 (2.94), 7.429 (1.47), 7.477 (0.62), 7.488 (1.54), 7.491 (1.60), 7.501 (5.13), 7.509 (4.63), 7.521 (12.78), 7.525 (1 1.60), 7.533 (6.95), 7.831 (2.36), 7.836 (2.20), 7.853 (3.67), 7.858 (3.58), 7.922 (7.1 1 ), 7.944 (4.32), 8.624 (1.49), 8.638 (2.12), 8.656 (1.47).
Trennung der Enantiomere:
Die Titelverbindung (137 mg) wurde in Methanol (17 ml) gelöst und mittels praparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 30 und 31 ) [Säule: Daicel Chiralpak AD, 250 mm x 20 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 1.0 ml; Eluent: 70% Kohlendioxid / 30% Methanol; Laufzeit 7 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet.
Beispiel 30
(+)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(2-fluorphenyl)-2-methyl- propansäure (Enantiomer 1)
Bei der in Beispiel 29 beschriebenen Enantiomerentrennung wurden 46 mg (97% Reinheit, ee-Wert 100%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = +71.4°, 589 nm, c = 0.37 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.03 min; MS (ESIpos): m/z = 521/523 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (0.62), 1.636 (16.00), 2.093 (1.40), 3.768 (1.33), 3.781 (1.46), 3.801 (1.60), 3.814 (1.48), 4.249 (1.59), 4.268 (1.74), 4.283 (1.54), 4.302 (1.43), 7.153 (1.34), 7.173 (1.85), 7.183 (1.86), 7.205 (3.32), 7.226 (1.68), 7.335 (0.83), 7.352 (1.42), 7.366 (1.32), 7.388 (2.03), 7.409 (2.90), 7.428 (1.48), 7.475 (0.62), 7.490 (1.52), 7.500 (4.83), 7.507 (4.32), 7.519 (12.55), 7.523 (1 1.56), 7.828 (2.08), 7.833 (2.00), 7.850 (3.30), 7.855 (3.33), 7.920 (6.27), 7.942 (3.91 ), 8.622 (1.47), 8.639 (2.12), 8.653 (1.46), 12.650 (2.52).
Beispiel 31
(-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(2-fluorphenyl)-2-methylpropan- säure (Enantiomer 2)
Bei der in Beispiel 29 beschriebenen Enantiomerentrennung wurden 56 mg (96% Reinheit, ee-Wert 100%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = -70.5°, 589 nm, c = 0.39 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.03 min; MS (ESIpos): m/z = 521/523 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.52), 0.008 (1.28), 1.030 (0.98), 1.045 (0.99), 1.283 (0.68), 1.636 (16.00), 2.092 (1.46), 2.522 (0.81 ), 3.768 (1.38), 3.780 (1.48), 3.801 (1.63), 3.814 (1.48), 4.249 (1.59), 4.267 (1.73), 4.282 (1.55), 4.301 (1.42), 7.152 (1.36), 7.173 (1 .90), 7.182 (1.95), 7.205 (3.39), 7.225 (1.73), 7.335 (0.87), 7.350 (1.46), 7.366 (1.37), 7.388 (2.08), 7.408 (2.98), 7.428 (1.53), 7.475 (0.67), 7.486 (1.54), 7.490 (1.62), 7.499 (5.05), 7.507 (4.63), 7.519 (12.83), 7.523 (1 1.68), 7.531 (7.29), 7.827 (2.24), 7.833 (2.09), 7.850 (3.52), 7.855 (3.45), 7.919 (6.36), 7.942 (3.96), 8.622 (1.51 ), 8.636 (2.17), 8.653 (1.48), 12.654 (2.75). Beispiel 32
(+/-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(2,6-difluorphenyl)-2-methyl- propansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 2-(2,6-difluorphenyl)-2-methylpropanoat (625 mg, 98% Reinheit, 1.03 mmol, Beispiel 163A) in Dichlormethan (7.3 ml) wurde TFA (1.6 ml, 21 mmol) gegeben, und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 20) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der erhaltene Rückstand im Vakuum getrocknet. Es wurden 492 mg (98% Reinheit, 87% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.01 min; MS (ESIpos): m/z = 539/541 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.84), 0.008 (1.73), 1.761 (12.93), 2.073 (2.77), 2.159 (3.62), 2.327 (0.40), 2.523 (1.15), 3.787 (1.42), 3.800 (1.55), 3.820 (1.75), 3.834 (1.59), 4.196 (1.88), 4.214 (2.04), 4.230 (1.79), 4.248 (1.62), 7.047 (3.1 1 ), 7.069 (5.05), 7.094 (3.52), 7.377 (1.49), 7.394 (2.01 ), 7.410 (1.37), 7.480 (1.03), 7.492 (2.99), 7.507 (8.37), 7.513 (6.86), 7.525 (13.93), 7.538 (16.00), 7.555 (3.1 1 ), 7.839 (3.67), 7.845 (3.43), 7.862 (5.71 ), 7.867 (5.61 ), 7.933 (10.68), 7.955 (6.66), 8.796 (2.23), 8.812 (3.84), 8.828 (2.16), 12.833 (0.42).
Trennung der Enantiomere:
Die Titelverbindung (450 mg) wurde in Isopropanol (5 ml) gelöst und mittels praparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 33 und 34) [Säule: Daicel Chiral- pak AF, 5 μηη, 250 mm x 20 mm; Fluss: 15 ml/min; Detektion: 220 nm; Temperatur: 55 °C; Injektion: 0.3 ml; Eluent: 50% Heptan / (50% Isopropanol + 0.2% Essigsäure); Laufzeit 10 min, isok- ratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert.
Beispiel 33
(+)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(2,6-difluorphenyl)-2-methyl- propansäure (Enantiomer 1)
Bei der in Beispiel 32 beschriebenen Enantiomerentrennung wurden 21 1 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = +62.7°, 589 nm, c = 0.25 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.92 min; MS (ESIpos): m/z = 539/541 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.762 (13.17), 2.073 (4.25), 2.160 (3.79), 3.788 (1.60), 3.801 (1.75), 3.821 (1.98), 3.835 (1.83), 4.197 (1.96), 4.215 (2.12), 4.231 (1.85), 4.249 (1.66), 7.047 (3.15), 7.070 (5.21 ), 7.094 (3.51 ), 7.376 (1.54), 7.393 (2.06), 7.41 1 (1.42), 7.480 (1.02), 7.492 (2.88), 7.507 (8.19), 7.513 (6.38), 7.525 (13.90), 7.537 (16.00), 7.555 (3.03), 7.629 (0.49), 7.839 (3.36), 7.844 (3.05), 7.861 (5.25), 7.867 (4.98), 7.933 (9.45), 7.955 (5.90), 8.797 (2.25), 8.813 (3.94), 8.829 (2.17), 12.831 (0.43).
Beispiel 34
(-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(2,6-difluorphenyl)-2-methyl- propansäure (Enantiomer 2)
Bei der in Beispiel 32 beschriebenen Enantiomerentrennung wurden 209 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = -50.8°, 589 nm, c = 0.42 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.92 min; MS (ESIpos): m/z = 539/541 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.761 (13.10), 2.073 (1.14), 2.159 (3.74), 2.328 (0.45), 2.669 (0.41 ), 3.787 (1.42), 3.801 (1.54), 3.821 (1.75), 3.835 (1.60), 4.196 (1.84), 4.213 (2.00), 4.230 (1.78), 4.248 (1.62), 7.047 (3.13), 7.069 (5.21 ), 7.094 (3.54), 7.376 (1.52), 7.393 (2.05), 7.410 (1.40), 7.480 (0.96), 7.493 (2.78), 7.508 (8.25), 7.514 (6.48), 7.526 (14.06), 7.539 (16.00), 7.556 (3.14), 7.841 (3.42), 7.846 (3.18), 7.864 (5.38), 7.869 (5.24), 7.934 (10.00), 7.956 (6.16), 8.797 (2.20), 8.814 (3.90), 8.829 (2.18).
Beispiel 35
(+/-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(2-chlorphenyl)-2-methyl- propansäure (Racemat)
Zu einer Lösung aus (+/-)-Methyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2- (2-chlorphenyl)-2-methylpropanoat (540 mg, 978 μηηοΙ, Beispiel 164A) in THF (8.4 ml) und Methanol (120 μΙ) wurde bei RT 1 M Natronlauge (5.9 ml, 5.9 mmol) gegeben, und das Gemisch wurde 8 h bei 40 °C gerührt. Anschließend wurde erneut Natronlauge (5.9 ml, 5.9 mmol) hinzugegeben, und das Gemisch wurde weitere 56 h bei 40 °C gerührt. Anschließend wurde das Gemisch mit TFA auf pH 3 eingestellt und mittels praparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand wurde lyophilisiert. Es wurden 296 mg (98% Reinheit, 55% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.03 min; MS (ESIpos): m/z = 537/539 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.007 (1.18), 0.008 (0.90), 1.646 (1.14), 1.688 (14.53), 2.073 (1.84), 2.523 (0.85), 2.731 (5.71 ), 2.890 (7.16), 3.792 (1.39), 3.804 (1.61 ), 3.825 (1.65), 3.838 (1.60), 4.526 (1.74), 4.545 (1.94), 4.560 (1.78), 4.579 (1.65), 7.323 (1.71 ), 7.340 (3.30), 7.354 (2.56), 7.372 (1.08), 7.438 (2.79), 7.456 (5.35), 7.460 (4.71 ), 7.475 (3.07), 7.480 (2.90), 7.491 (1.83), 7.501 (5.16), 7.509 (5.14), 7.520 (16.00), 7.531 (6.71 ), 7.830 (2.22), 7.835 (2.1 1 ), 7.852 (3.59), 7.857 (3.57), 7.918 (6.86), 7.931 (0.60), 7.941 (4.22), 7.952 (1.10), 8.563 (1.62), 8.576 (2.18), 8.581 (2.18), 8.595 (1.59).
Trennung der Enantiomere:
Die Titelverbindung (275 mg) wurde in Ethanol (10 ml) gelöst und mittels praparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 36 und 37) [Säule: YMC Chiralart Amylose SA, 5 μηη, 250 mm x 30 mm; Fluss: 30 ml/min; Detektion: 220 nm; Temperatur: 40 °C; Injektion: 0.5 ml; Eluent: 50% Heptan / (50% Ethanol + 0.2% Essigsäure); Laufzeit 9.5 min, isok- ratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand wurde lyophilisiert.
Beispiel 36
(-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(2-chlorphenyl)-2-methyl- propansäure (Enantiomer 1)
Bei der in Beispiel 35 beschriebenen Enantiomerentrennung wurde die vorgereinigte Titelverbindung als früher eluierendes Enantiomer erhalten (ee-Wert 99%) und mittels präparativer HPLC (Methode 15) nachgereinigt. Es wurden 68 mg (98% Reinheit) der Titelverbindung erhalten.
[a]D 20 = -94.8°, 589 nm, c = 0.40 g/100 ml, Chloroform
LC-MS (Methode 2): Rt = 1.03 min; MS (ESIpos): m/z = 357/539 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (3.09), 0.008 (2.81 ), 1.294 (0.51 ), 1.645 (0.42), 1.687 (14.26), 2.073 (1.20), 2.328 (0.54), 2.366 (0.43), 2.518 (2.16), 2.523 (1.63), 2.670 (0.50), 3.790 (1.44), 3.803 (1.59), 3.824 (1.70), 3.837 (1.59), 4.395 (0.60), 4.526 (2.20), 4.544 (2.37), 4.559 (2.19), 4.579 (2.04), 7.323 (1.68), 7.340 (3.24), 7.354 (2.51 ), 7.372 (1.03), 7.437 (2.65), 7.456 (5.25), 7.460 (4.68), 7.474 (3.08), 7.480 (2.87), 7.487 (1.69), 7.490 (1 .78), 7.500 (5.04), 7.508 (5.09), 7.519 (16.00), 7.530 (6.41 ), 7.533 (5.70), 7.828 (2.22), 7.834 (2.09), 7.851 (3.55), 7.856 (3.52), 7.917 (6.88), 7.939 (4.19), 8.561 (1.60), 8.575 (2.13), 8.580 (2.12), 8.593 (1.54).
Beispiel 37
(+)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(2-chlorphenyl)-2-methyl- propansäure (Enantiomer 2)
Bei der in Beispiel 35 beschriebenen Enantiomerentrennung wurde die vorgereinigte Titelverbindung als später eluierendes Enantiomer erhalten (ee-Wert 99%) und mittels präparativer HPLC (Methode 15) nachgereinigt. Es wurden 99 mg (98% Reinheit) der Titelverbindung erhalten.
[a]D 20 = +96.8°, 589 nm, c = 0.33 g/100 ml, Chloroform
LC-MS (Methode 2): Rt = 1.03 min; MS (ESIpos): m/z = 537/539 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.30), 0.008 (1.81 ), 1.645 (1.12), 1.687 (14.19), 2.072 (1.44), 2.522 (1.04), 3.790 (1.48), 3.803 (1.68), 3.824 (1.74), 3.837 (1.71 ), 4.174 (0.72), 4.191 (0.70), 4.207 (0.68), 4.226 (0.65), 4.525 (1.83), 4.544 (1.97), 4.559 (1 .83), 4.578 (1.68), 7.322 (1.70), 7.339 (3.25), 7.353 (2.52), 7.371 (1.06), 7.437 (2.76), 7.456 (5.26), 7.459 (4.65), 7.474 (3.03), 7.479 (2.82), 7.489 (1.82), 7.500 (5.03), 7.508 (5.01 ), 7.518 (16.00), 7.530 (6.62), 7.828 (2.17), 7.833 (2.03), 7.850 (3.52), 7.855 (3.41 ), 7.917 (6.47), 7.929 (0.56), 7.939 (3.95), 8.561 (1.58), 8.575 (2.15), 8.579 (2.13), 8.593 (1.54).
Beispiel 38
(+/-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-fluor-2-phenylpropansäure (Racemat)
Zu einer Lösung aus (+/-)-Ethyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2- fluorhydratropaat (196 mg, 366 μηηοΙ, Beispiel 165A) in THF (3.0 ml) und Methanol (1.5 ml) wurde bei RT 1 M Natronlauge (2.2 ml, 2.2 mmol) gegeben, und das Gemisch wurde 2.5 h bei RT gerührt. Anschließend wurde das Gemisch mit TFA (200 μΙ, 2.6 mmol) auf pH 3 eingestellt und mittels präparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand wurde im Vakuum getrocknet. Es wurden 167 mg (100% Reinheit, 90% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 0.95 min; MS (ESIpos): m/z = 507/509 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 2.085 (1.43), 2.149 (1.02), 2.327 (0.60), 2.347 (0.53), 3.169 (1.97), 3.184 (1.79), 4.255 (1.04), 4.297 (0.88), 4.351 (0.88), 4.369 (0.93), 4.389 (0.69), 4.413 (1.13), 4.430 (0.99), 4.450 (0.65), 4.466 (0.59), 5.754 (0.61 ), 7.383 (0.77), 7.392 (0.91 ), 7.460 (4.35), 7.477 (6.02), 7.488 (5.13), 7.502 (8.20), 7.509 (7.36), 7.522 (16.00), 7.583 (9.85), 7.601 (7.29), 7.827 (2.75), 7.849 (4.09), 7.926 (8.71 ), 7.949 (5.53), 9.1 18 (2.79), 13.835 (1.53). Trennung der Enantiomere:
Die Titelverbindung (140 mg) wurde in 15 ml eines Methanol-Acetonitril-Gemisches gelöst und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 39 und 40) [Säule: Daicel Chiralpak AD, 250 mm x 20 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 2.0 ml; Eluent: 75% Kohlendioxid / 25% Isopropanol; Laufzeit 8 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet.
Beispiel 39
(-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-fluor-2-phenylpropansäure (Enantiomer 1)
Bei der in Beispiel 38 beschriebenen Enantiomerentrennung wurden 58 mg (95% Reinheit, ee-Wert 100%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -27.0°, 589 nm, c = 0.38 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.77 min; MS (ESIpos): m/z = 507/509 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.19), 0.008 (1.92), 2.1 19 (0.93), 2.263 (1.83), 2.327 (0.73), 2.366 (0.41 ), 2.429 (0.68), 2.669 (0.47), 4.200 (0.52), 4.238 (0.96), 4.280 (0.77), 4.353 (0.80), 4.370 (0.83), 4.389 (0.64), 4.414 (1.03), 4.432 (0.89), 4.451 (0.62), 4.468 (0.55), 7.453 (4.07), 7.472 (5.44), 7.488 (4.67), 7.502 (7.86), 7.509 (6.85), 7.522 (16.00), 7.562 (1.88), 7.580 (9.87), 7.598 (7.25), 7.826 (2.75), 7.830 (2.82), 7.848 (3.96), 7.852 (3.89), 7.863 (0.90), 7.886 (0.77), 7.891 (0.73), 7.924 (8.83), 7.947 (5.69), 7.963 (1.16), 7.985 (0.86), 9.102 (2.47), 9.170 (0.44), 13.870 (0.52). Beispiel 40
(+)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-fluor-2-phenylpropansäure (Enantiomer 2)
Bei der in Beispiel 38 beschriebenen Enantiomerentrennung wurden 41 mg (100% Reinheit, ee-Wert 100%) der Titelverbindung als später eluierendes Enantiomer erhalten. [a]D 20 = +26.2°, 589 nm, c = 0.40 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.77 min; MS (ESIpos): m/z = 507/509 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 2.148 (1.06), 2.327 (0.82), 2.668 (0.55), 4.238 (1.10), 4.283 (0.84), 4.370 (0.89), 4.414 (1.03), 7.454 (4.26), 7.473 (5.72), 7.488 (4.83), 7.502 (7.38), 7.509 (6.94), 7.522 (16.00), 7.580 (9.88), 7.599 (7.46), 7.826 (2.84), 7.848 (4.32), 7.924 (9.20), 7.947 (5.83), 9.103 (2.71 ), 13.871 (0.63).
Beispiel 41
(+/-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-methyl-2-[3-(trifluormethyl)- phenyl]propansäure (Racemat)
Zu einer Lösung aus (+/-)-Ethyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2- methyl-2-[3-(trifluormethyl)phenyl]propanoat (550 mg, 939 mol, Beispiel 166A) in THF (8.0 ml) und Methanol (4.0 ml) wurde bei RT 1 M Natronlauge (5.6 ml, 5.6 mmol) gegeben, und das Gemisch wurde 24 h bei RT gerührt. Anschließend wurde das Gemisch mit TFA (510 μΙ, 6.6 mmol) auf pH 3 eingestellt und mittels präparativer HPLC (Methode 20) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand wurde im Vakuum getrocknet. Es wurden 480 mg (98% Reinheit, 88% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.10 min; MS (ESIpos): m/z = 571/573 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.47), 0.008 (0.41 ), 1.693 (15.91 ), 2.040 (0.83), 2.073 (0.93), 3.652 (0.97), 3.817 (1.22), 3.829 (1.29), 3.851 (1.44), 3.863 (1.30), 4.293 (0.94), 4.31 1 (1.01 ), 4.326 (0.92), 4.345 (0.81 ), 7.486 (1.18), 7.499 (3.81 ), 7.506 (4.35), 7.516 (16.00), 7.620 (0.98), 7.639 (2.39), 7.658 (2.41 ), 7.675 (2.77), 7.694 (1.30), 7.727 (4.34), 7.761 (2.69), 7.780 (1.95), 7.826 (1.86), 7.831 (1.75), 7.848 (2.83), 7.853 (2.78), 7.925 (5.80), 7.947 (3.73), 8.700 (1.33), 8.713 (1.87), 8.717 (1.87), 8.730 (1.32).
Trennung der Enantiomere:
Die Titelverbindung (430 mg) wurde in 20 ml eines Methanol-Acetonitril-Gemsches gelöst und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 42 und 43) [Säule: Daicel Chiralpak AD, 250 mm x 20 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 0.8 ml; Eluent: 82% Kohlendioxid / 18% Ethanol; Laufzeit 10 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet.
Beispiel 42
(-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-methyl-2-[3-(trifluormethyl)- phenyljpropansäure (Enantiomer 1)
Bei der in Beispiel 41 beschriebenen Enantiomerentrennung wurde die vorgereinigte Titelverbin- dung als früher eluierendes Enantiomer (ee-Wert 100%) erhalten und mittels präparativer HPLC (Methode 20) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt und lyophilisiert. Es wurden 180 mg (98% Reinheit) der Titelverbindung erhalten.
[a]D 20 = -36.7°, 589 nm, c = 0.40 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.08 min; MS (ESIpos): m/z = 571/573 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.195 (0.51 ), 1.21 1 (0.52), 1.694 (15.62), 2.042 (0.84), 2.073 (0.85), 3.819 (1.17), 3.831 (1.22), 3.853 (1.38), 3.865 (1.24), 4.294 (1.09), 4.31 1 (1.14), 4.327 (1.03), 4.347 (0.92), 7.499 (4.1 1 ), 7.507 (4.61 ), 7.517 (16.00), 7.621 (1.03), 7.640 (2.46), 7.659 (2.45), 7.675 (2.86), 7.695 (1.30), 7.728 (4.32), 7.762 (2.63), 7.781 (1.95), 7.827 (2.02),
7.832 (1.88), 7.849 (3.02), 7.854 (2.93), 7.926 (6.33), 7.948 (3.99), 8.701 (1.33), 8.714 (1.83), 8.732 (1.24).
Beispiel 43
(+)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-m
phenyl]propansäure (Enantiomer 2)
Bei der in Beispiel 41 beschriebenen Enantiomerentrennung wurde die vorgereinigte Titelverbindung als später eluierendes Enantiomer (ee-Wert 99%) erhalten und mittels präparativer HPLC (Methode 20) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt und lyophilisiert. Es wurden 164 mg (98% Reinheit) der Titelverbindung erhalten. [a]D 20 = +30.4°, 589 nm, c = 0.43 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.08 min; MS (ESIpos): m/z = 571/573 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.61 ), 0.008 (0.58), 1.693 (16.00), 2.038 (0.80), 2.523 (0.55), 3.818 (1.10), 3.830 (1.19), 3.852 (1.36), 3.864 (1.22), 4.293 (0.93), 4.31 1 (1.02), 4.327 (0.93), 4.345 (0.81 ), 7.474 (0.41 ), 7.488 (1.20), 7.501 (4.00), 7.508 (4.56), 7.518 (15.96), 7.531 (4.1 1 ), 7.621 (0.98), 7.639 (2.40), 7.659 (2.40), 7.676 (2.70), 7.695 (1.20), 7.728 (4.31 ), 7.761 (2.62), 7.780 (1.92), 7.828 (1.90), 7.834 (1.77), 7.851 (2.93), 7.856 (2.84), 7.927 (6.01 ), 7.949 (3.84), 8.702 (1.33), 8.715 (1.86), 8.719 (1.84), 8.732 (1.29).
Beispiel 44
(+/-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(3-methoxyphenyl)propan- säure (Racemat)
Zu einer Suspension aus (+/-)-ie/f-Butyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]- amino}-2-(3-methoxyphenyl)propanoat (940 mg, 1.63 mmol, Beispiel 167A) in Dichlormethan (26 ml) wurde bei RT TFA (13 ml, 160 mmol) gegeben, und das Gemisch wurde 16 h bei RT ge- rührt. Anschließend wurde das Gemisch eingeengt, und der Rückstand wurde mehrmals hinter-
einander mit Dichlormethan versetzt und wieder eingeengt. Anschließend wurde der Rückstand im Vakuum getrocknet. Es wurden 1.01 g (99% Reinheit,„>100%" d. Th., TFA- bzw. lösungsmit- telhaltig) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.85 min; MS (ESIpos): m/z = 519/521 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.149 (0.58), 0.008 (5.19), 0.147 (0.63), 2.160 (5.22), 2.327 (1.01 ), 2.366 (0.56), 2.670 (1.08), 3.760 (1.54), 3.826 (1.78), 3.845 (3.51 ), 3.861 (3.56), 3.877 (2.29), 3.958 (2.97), 3.977 (4.16), 3.996 (1.64), 5.176 (0.79), 6.875 (2.53), 6.896 (2.92), 6.924 (5.75), 6.947 (3.63), 6.966 (3.86), 7.278 (3.41 ), 7.298 (5.40), 7.317 (2.76), 7.509 (5.61 ), 7.516 (5.28), 7.528 (16.00), 7.757 (1.29), 7.844 (3.1 1 ), 7.849 (2.72), 7.867 (4.66), 7.872 (4.38), 7.940 (8.61 ), 7.963 (5.33), 8.948 (1.73), 8.962 (3.23), 8.977 (1.64).
Trennung der Enantiomere:
Die Titelverbindung (400 mg) wurde in einem Gemisch aus Ethanol, Methanol und Acetonitril (jeweils 5 ml) in der Wärme gelöst und mittels präparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 45 und 46) [Säule: Daicel Chiralpak ID, 5 μηη, 250 mm x 20 mm; Fluss: 15 ml/min; Detektion: 210 nm; Temperatur: 25 °C; Injektion: 0.50 ml; Eluent: 50% Heptan / (50% Ethanol + 0.2% Essigsäure + 1 % Wasser); Laufzeit 18 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand wurde in Acetonitril/Wasser lyophilisiert.
Beispiel 45
(-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(3-methoxyphenyl)propansäure (Enantiomer 1)
Bei der in Beispiel 44 beschriebenen Enantiomerentrennung wurden 122 mg (95% Reinheit, ee-Wert 99%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -39.2°, 589 nm, c = 0.44 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.86 min; MS (ESIpos): m/z = 519/529 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.030 (1.19), 1.045 (1.18), 1.154 (0.57), 1.375 (0.48), 2.159 (6.15), 2.213 (1.48), 3.313 (8.74), 3.723 (2.36), 3.792 (0.68), 3.812 (1.00), 3.825 (2.18), 3.844 (3.99), 3.860 (4.09), 3.875 (2.49), 3.895 (0.83), 3.908 (0.70), 3.922 (0.42), 3.955 (3.1 1 ), 3.974 (4.32), 3.994 (1.63), 6.873 (2.90), 6.893 (3.29), 6.923 (6.09), 6.947 (3.91 ), 6.966 (4.03), 7.276 (3.32), 7.296 (5.16), 7.315 (2.59), 7.481 (1.36), 7.495 (2.74), 7.506 (6.47), 7.513 (6.14), 7.526 (16.00), 7.529 (15.41 ), 7.756 (1.44), 7.839 (2.93), 7.844 (2.59), 7.861 (4.27), 7.867 (3.96), 7.937 (7.45), 7.959 (4.74), 8.944 (1.97), 8.958 (3.49), 8.971 (1.77), 12.609 (0.55).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.61 (br. s, 1 H), 8.96 (t, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.76 (br. s, 1 H), 7.59-7.40 (m, 5H), 7.30 (t, 1 H), 7.01-6.83 (m, 3H), 4.03-3.93 (m, 1 H), 3.92- 3.77 (m, 2H), 3.74 (s, 3H), 2.16 (br. s, 3H).
Beispiel 46
(+)-3-{[(6-Brom-3-methyl-2-phenylcN^
säure (Enantiomer 1)
Bei der in Beispiel 44 beschriebenen Enantiomerentrennung wurden 123 mg (95% Reinheit, ee-Wert 98%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = +49.0°, 589 nm, c = 0.37 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.86 min; MS (ESIpos): m/z = 519/521 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (1.50), 1.030 (3.72), 1.045 (3.75), 1.156 (0.53), 1.283 (0.56), 1.909 (0.81 ), 2.159 (5.77), 2.213 (1.18), 2.523 (1.58), 3.313 (1 1.20), 3.650 (0.45), 3.723 (1.63), 3.792 (0.55), 3.812 (0.80), 3.825 (1.95), 3.845 (3.74), 3.861 (3.87), 3.876 (2.36), 3.896 (0.75), 3.909 (0.60), 3.957 (2.87), 3.976 (4.15), 3.995 (1.55), 6.869 (2.25), 6.874 (2.64), 6.889 (2.52), 6.895 (3.00), 6.923 (5.90), 6.947 (3.79), 6.966 (4.00), 7.277 (3.34), 7.296 (5.24), 7.316 (2.69), 7.481 (0.79), 7.493 (1.86), 7.496 (2.08), 7.506 (5.87), 7.514 (5.67), 7.526 (16.00), 7.529 (15.28), 7.538 (8.90), 7.756 (1.29), 7.840 (2.99), 7.845 (2.60), 7.862 (4.46), 7.868 (4.12), 7.938 (7.77), 7.960 (5.00), 8.945 (1.80), 8.958 (3.36), 8.972 (1.72), 12.595 (0.45).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.59 (br. s, 1 H), 8.96 (t, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.76 (br. s, 1 H), 7.58-7.47 (m, 5H), 7.30 (t, 1 H), 7.00-6.85 (m, 3H), 4.02-3.94 (m, 1 H), 3.92- 3.78 (m, 2H), 3.75 (s, 3H), 2.16 (br. s, 3H).
Beispiel 47
(+/-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(3-chlorphenyl)propansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 2-(3-chlorphenyl)propanoat (430 mg, 741 μηηοΙ, Beispiel 168A) in Dichlormethan (12 ml) wurde TFA (5.7 ml, 74 mmol) gegeben, und das Gemisch wurde 16 h bei RT gerührt. Anschließend
wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde im Vakuum getrocknet. Es wurden 420 mg (96% Reinheit,„>100%" d. Th., TFA- bzw. lösungsmittelhaltig) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.97 min; MS (ESIpos): m/z = 523/525 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.98), 0.008 (2.34), 2.134 (3.31 ), 2.328 (0.48), 2.670 (0.49), 3.878 (2.54), 3.896 (4.45), 3.91 1 (3.63), 4.017 (2.62), 4.036 (3.82), 4.056 (1.58), 5.754 (0.64), 7.358 (2.19), 7.376 (5.07), 7.394 (4.03), 7.406 (5.31 ), 7.424 (3.24), 7.438 (6.04), 7.483 (0.52), 7.497 (1.49), 7.507 (4.79), 7.515 (4.57), 7.526 (16.00), 7.537 (7.13), 7.703 (0.59), 7.842 (2.44), 7.848 (2.23), 7.865 (3.83), 7.870 (3.74), 7.937 (7.12), 7.959 (4.51 ), 8.944 (1.58), 8.958 (3.38), 8.972 (1.65).
Trennung der Enantiomere:
Die Titelverbindung (350 mg) wurde in einem Gemisch aus Ethanol, Methanol und Acetonitril (jeweils 5 ml) in der Wärme gelöst und mittels praparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 48 und 49) [Säule: Daicel Chiralpak AD-H, 5 μηη, 250 mm x 20 mm; Fluss: 15 ml/min; Detektion: 210 nm; Temperatur: 25 °C; Injektion: 0.50 ml; Eluent: 40% Heptan / (60% Ethanol + 0.2% Essigsäure + 1% Wasser); Laufzeit 25 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand wurde in Aceton itril/Wasser lyophilisiert.
Beispiel 48
(-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(3-chlorphenyl)propansäure (Enantiomer 1)
Bei der in Beispiel 47 beschriebenen Enantiomerentrennung wurden 124 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -47.8°, 589 nm, c = 0.35 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.04 min; MS (ESIpos): m/z = 523/525 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.80), 0.008 (0.84), 1.140 (0.42), 1.158 (0.83), 1.177 (0.45), 1.234 (0.68), 1.909 (0.61 ), 2.135 (3.30), 2.188 (0.80), 2.269 (0.47), 2.523 (1.08), 2.788 (0.47), 3.182 (0.45), 3.878 (2.56), 3.897 (4.40), 3.91 1 (3.57), 4.018 (2.57), 4.036 (3.71 ), 4.056 (1.54), 7.358 (2.47), 7.362 (1.62), 7.376 (5.32), 7.380 (4.48), 7.394 (4.20), 7.406 (5.47), 7.424 (3.37), 7.438 (6.1 1 ), 7.471 (0.46), 7.482 (0.72), 7.495 (1.84), 7.505 (5.22), 7.513 (5.03), 7.524 (16.00), 7.532 (9.04), 7.536 (7.10), 7.539 (6.31 ), 7.547 (1.52), 7.701 (0.63), 7.839 (2.61 ), 7.844 (2.37), 7.861 (4.00), 7.867 (3.82), 7.935 (7.41 ), 7.957 (4.72), 8.943 (1.63), 8.957 (3.35), 8.971 (1.56), 12.729 (0.57).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.73 (br. s, 1 H), 8.96 (t, 1 H), 7.94 (d, 1 H), 7.86 (dd, 1 H), 7.70 (br. s, 1 H), 7.57-7.46 (m, 5H), 7.45-7.33 (m, 4H), 4.03 (t, 1 H), 3.90 (t, 2H), 2.14 (br. s, 3H).
Beispiel 49
(+)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2^
(Enantiomer 2)
Bei der in Beispiel 47 beschriebenen Enantiomerentrennung wurden 106 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = +46.2°, 589 nm, c = 0.34 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.04 min; MS (ESIpos): m/z = 523/525 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.140 (0.54), 1.158 (1.07), 1.176 (0.63), 1.234 (1.23), 2.134 (3.40), 2.188 (0.86), 2.327 (0.41 ), 2.669 (0.41 ), 3.877 (2.52), 3.895 (4.35), 3.910 (3.44), 4.016 (2.32), 4.035 (3.40), 4.054 (1.39), 7.357 (2.29), 7.375 (5.14), 7.393 (4.06), 7.405 (5.13), 7.424 (3.26), 7.437 (5.99), 7.481 (0.70), 7.495 (1.75), 7.505 (4.91 ), 7.512 (4.73), 7.524 (16.00), 7.535 (6.83), 7.700 (0.66), 7.839 (2.38), 7.844 (2.16), 7.861 (3.63), 7.866 (3.43), 7.934 (6.61 ), 7.957 (4.15), 8.942 (1.59), 8.956 (3.16), 8.969 (1.48), 12.665 (0.42).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.67 (br. s, 1 H), 8.96 (t, 1 H), 7.94 (d, 1 H), 7.86 (dd, 1 H), 7.70 (br. s, 1 H), 7.57-7.47 (m, 5H), 7.46-7.33 (m, 4H), 4.03 (t, 1 H), 3.90 (t, 2H), 2.13 (br. s, 3H).
Beispiel 50
(+/-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(3-methylphenyl)propansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 2-(3-methylphenyl)propanoat (830 mg, 1.48 mmol, Beispiel 169A) in Dichlormethan (24 ml) wurde TFA (11 ml, 150 mmol) gegeben, und das Gemisch wurde 16 h bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde im Vakuum getrocknet. Es wurden 920 mg (92% Rein- heit,„>100%" d. Th., TFA- bzw. lösungsmittelhaltig) der Titelverbindung erhalten.
LC-MS (Methode 2): R
t = 1.03 min; MS (ESIpos): m/z = 503/505 [M+H]
+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (1.20), 2.167 (3.60), 2.303 (16.00), 2.327 (0.50), 2.523 (1.18), 3.782 (0.63), 3.797 (1.15), 3.814 (1.25), 3.831 (0.97), 3.846 (0.81 ), 3.864 (1.13), 3.878 (1.33), 3.898 (0.70), 3.910 (0.64), 3.940 (2.04), 3.959 (2.94), 3.978 (1.10), 4.359 (1.54), 7.1 12 (1.44), 7.131 (1.85), 7.161 (1.65), 7.189 (3.89), 7.247 (2.1 1 ), 7.265 (3.01 ), 7.284 (1.15), 7.498 (0.97), 7.508 (3.33), 7.515 (2.94), 7.528 (8.62), 7.532 (8.10), 7.540 (4.93), 7.752 (0.56), 7.846 (1.70), 7.851 (1.50), 7.868 (2.62), 7.873 (2.47), 7.940 (4.71 ), 7.962 (2.96), 8.943 (1.05), 8.957 (2.00), 8.971 (1.04).
Trennung der Enantiomere:
Die Titelverbindung (400 mg) wurde in einem Gemisch aus Ethanol, Methanol und Acetonitril (jeweils 5 ml) in der Wärme gelöst und mittels präparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 51 und 52) [Säule: Daicel Chiralpak AD-H, 5 μηη, 250 mm x 20 mm; Fluss: 15 ml/min; Detektion: 210 nm; Temperatur: 25 °C; Injektion: 0.50 ml; Eluent: 30% Heptan / (70% Ethanol + 0.2% Essigsäure + 1% Wasser); Laufzeit 26 min, isokratisch]. Die vereinigten Ziel- fraktionen wurden eingeengt, und der Rückstand wurde in Acetonitril/Wasser lyophilisiert.
Beispiel 51
(-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(3-methylphenyl)propansäure (Enantiomer 1)
Bei der in Beispiel 50 beschriebenen Enantiomerentrennung wurden 131 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -53.2°, 589 nm, c = 0.33 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.03 min; MS (ESIpos): m/z = 503/505 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (0.44), 1.908 (0.56), 2.167 (3.72), 2.222 (0.42), 2.303 (16.00), 2.523 (0.60), 3.782 (0.58), 3.797 (1.07), 3.814 (1.15), 3.831 (0.87), 3.845 (0.72), 3.864 (1.03), 3.877 (1.20), 3.896 (0.58), 3.909 (0.55), 3.938 (1.86), 3.957 (2.71 ), 3.976 (0.92), 7.1 1 1 (1.46), 7.129 (1.89), 7.161 (1.67), 7.189 (3.87), 7.245 (2.14), 7.264 (2.99), 7.283 (1.16), 7.481 (0.44), 7.496 (1.04), 7.506 (3.39), 7.513 (3.00), 7.526 (8.50), 7.531 (7.97), 7.538 (5.1 1 ), 7.550 (1.10), 7.751 (0.60), 7.842 (1.66), 7.847 (1.46), 7.864 (2.54), 7.869 (2.37), 7.938 (4.63), 7.960 (2.95), 8.940 (1.05), 8.954 (1.95), 8.968 (1.01 ).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.60 (br. s, 1 H), 8.95 (t, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.75 (br. s, 1 H), 7.58-7.46 (m, 5H), 7.26 (t, 1 H), 7.22-7.08 (m, 3H), 4.01-3.73 (m, 3H), 2.30 (s, 3H), 2.17 (br. s, 3H).
Beispiel 52
(+)-3-{[(6-Brom-3-methyl-2-phenylcN^
(Enantiomer 2)
Bei der in Beispiel 50 beschriebenen Enantiomerentrennung wurden 1 19 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = +53.0°, 589 nm, c = 0.39 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.04 min; MS (ESIpos): m/z = 503/505 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.54), 0.008 (0.51 ), 1.235 (0.42), 2.166 (3.68), 2.303 (16.00), 2.522 (0.63), 3.782 (0.56), 3.796 (1.05), 3.814 (1.14), 3.830 (0.86), 3.845 (0.71 ), 3.864 (1.03), 3.877 (1.20), 3.897 (0.57), 3.909 (0.53), 3.939 (1.82), 3.958 (2.69), 3.976 (0.91 ), 7.1 1 1 (1.46), 7.129 (1.86), 7.161 (1.64), 7.188 (3.81 ), 7.245 (2.14), 7.264 (3.00), 7.283 (1.16), 7.480 (0.47), 7.492 (1.02), 7.495 (1.04), 7.506 (3.33), 7.513 (2.98), 7.525 (8.39), 7.530 (7.87), 7.538 (4.96), 7.550 (0.96), 7.747 (0.57), 7.841 (1.72), 7.846 (1.49), 7.863 (2.61 ), 7.869 (2.43), 7.937 (4.76), 7.960 (3.01 ), 8.940 (1.06), 8.953 (1.97), 8.967 (1.00).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.53 (br. s, 1 H), 8.95 (t, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.75 (br. s, 1 H), 7.58-7.46 (m, 5H), 7.26 (t, 1 H), 7.21 -7.08 (m, 3H), 4.01-3.74 (m, 3H), 2.30 (s, 3H), 2.17 (br. s, 3H).
Beispiel 53
(+/-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-hydroxy-2-(2-methoxy- phenyl)propansäure (Racemat)
Zu einer Lösung aus (+/-)-Methyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2- hydroxy-2-(2-methoxyphenyl)propanoat (220 mg, 400 μηιοΙ, Beispiel 170A) in THF (3.4 ml) und Methanol (680 μΙ) wurde 1 M Natronlauge (2.4 ml, 2.4 mmol) gegeben, und das Gemisch wurde 4.5 h bei RT gerührt. Anschließend wurde das Gemisch mit TFA (220 μΙ, 2.8 mmol) auf pH 2-3
eingestellt und mittels präparativer HPLC (Methode 15) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde in Aceton itril/Wasser lyophilisiert. Es wurden 202 mg (96% Reinheit, 91 % d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 0.93 min; MS (ESIpos): m/z = 535/537 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.70), 0.008 (1.71 ), 2.072 (3.41 ), 2.391 (0.68), 2.523 (0.85), 3.952 (0.89), 3.985 (1.08), 4.271 (0.86), 6.925 (1.75), 6.944 (3.55), 6.962 (2.03), 6.987 (2.93), 7.007 (3.32), 7.261 (1.33), 7.280 (2.1 1 ), 7.299 (1.08), 7.473 (0.64), 7.483 (1.54), 7.486 (1.71 ), 7.496 (4.98), 7.504 (4.81 ), 7.516 (16.00), 7.523 (8.90), 7.527 (7.34), 7.595 (2.60), 7.614 (2.38), 7.816 (2.42), 7.821 (2.23), 7.838 (3.87), 7.843 (3.77), 7.898 (7.48), 7.921 (4.40), 8.410 (1.49), 8.424 (2.08), 8.440 (1.37).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.40 (br. s, 1 H), 8.43 (t, 3H), 7.91 (d, 1 H), 7.83 (dd, 1 H), 7.69 (br. s, 1 H), 7.61 (d, 1 H), 7.55-7.46 (m, 5H), 7.28 (t, 1 H), 7.06-6.88 (m, 2H), 6.41 (br. s, 1 H), 4.35-4.20 (m, 1 H), 4.02-3.87 (m, 1 H), 3.77 (s, 3H), 2.07 (br. s, 3H).
Beispiel 54
(-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-hydroxy-2-(2-methoxyphenyl)- propansäure (Enantiomer 1)
Zu einer Lösung aus (-)-Methyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2- hydroxy-2-(2-methoxyphenyl)propanoat (128 mg, 233 μηιοΙ, Beispiel 171A) in THF (2.0 ml) und Methanol (500 μΙ) wurde 1 M Natronlauge (1.4 ml, 1.4 mmol) gegeben, und das Gemisch wurde 4.5 h bei RT gerührt. Anschließend wurde das Gemisch mit TFA (130 μΙ, 1.6 mmol) auf pH 2-3 eingestellt und mittels präparativer HPLC (Methode 15) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 120 mg (97% Reinheit, ee-Wert 100%, 94% d. Th.) der Titelverbindung erhalten.
[a]D 20 = -72.0°, 589 nm, c = 0.44 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 0.93 min; MS (ESIpos): m/z = 535/537 [M+H]+
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: -0.007 (0.79), 2.075 (3.30), 2.084 (3.40), 2.392 (0.65), 2.518 (0.53), 3.918 (0.70), 3.954 (0.92), 3.985 (1.1 1 ), 4.276 (1.10), 5.752 (1.38), 6.930 (2.20), 6.945 (4.59), 6.960 (2.58), 6.989 (3.67), 7.005 (4.06), 7.266 (1.49), 7.280 (2.47), 7.295 (1.31 ), 7.475 (0.99), 7.484 (2.53), 7.489 (1.94), 7.496 (5.04), 7.499 (4.90), 7.501 (6.26), 7.515 (16.00), 7.520 (15.75), 7.525 (15.74), 7.600 (3.13), 7.615 (2.99), 7.674 (0.42), 7.818 (3.40), 7.822 (3.23), 7.836 (5.00), 7.840 (4.99), 7.900 (10.21 ), 7.918 (6.69), 8.414 (1.89), 8.426 (2.86), 8.438 (1.97).
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: 12.40 (br. s, 1 H), 8.43 (t, 1 H), 7.91 (d, 1 H), 7.83 (dd, 1 H), 7.67 (br. s, 1 H), 7.61 (d, 1 H), 7.56-7.44 (m, 5H), 7.28 (t, 1 H), 7.03-6.90 (m, 2H), 5.95 (br. s, 1 H), 4.33-4.20 (m, 1 H), 4.01-3.90 (m, 1 H), 3.78 (s, 3H), 2.07 (br. s, 3H).
Beispiel 55
(+)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-h
propansäure (Enantiomer 2)
Zu einer Lösung aus (+)-Methyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2- hydroxy-2-(2-methoxyphenyl)propanoat (120 mg, 219 μmol, Beispiel 172A) in THF (1.9 ml) und Methanol (470 μΙ) wurde 1 M Natronlauge (1.3 ml, 1.3 mmol) gegeben, und das Gemisch wurde 4.5 h bei RT gerührt. Anschließend wurde das Gemisch mit TFA (120 μΙ, 1.5 mmol) auf pH 2-3 eingestellt und mittels präparativer HPLC (Methode 15) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 1 1 1 mg (96% Reinheit, ee-Wert 99%, 91 % d. Th.) der Titelverbindung erhalten.
[a]D 20 = +71.7°, 589 nm, c = 0.41 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 0.93 min; MS (ESIpos): m/z = 535/537 [M+H]+
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: -0.006 (0.57), 2.085 (16.00), 2.392 (0.51 ), 2.518 (0.41 ), 3.918 (0.50), 3.949 (0.64), 3.985 (0.83), 4.275 (0.78), 5.752 (0.88), 6.931 (1.61 ), 6.945 (3.28), 6.961 (1.85), 6.989 (2.65), 7.006 (2.89), 7.265 (1.10), 7.280 (1.77), 7.295 (0.95), 7.476 (0.77), 7.485 (1.85), 7.490 (1.46), 7.497 (3.71 ), 7.500 (3.61 ), 7.502 (4.50), 7.516 (1 1.56), 7.521 (1 1.31 ), 7.526 (1 1.01 ), 7.600 (2.23), 7.614 (2.12), 7.820 (2.41 ), 7.824 (2.30), 7.838 (3.56), 7.842 (3.54), 7.902 (7.29), 7.920 (4.74), 8.415 (1.36), 8.427 (2.04), 8.439 (1.38).
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: 12.50 (br. s, 1 H), 8.43 (t, 1 H), 7.91 (d, 1 H), 7.83 (dd, 1 H), 7.68 (br. s, 1 H), 7.61 (d, 1 H), 7.55-7.46 (m, 5H), 7.28 (t, 1 H), 7.04-6.90 (m, 2H), 5.94 (br. s, 1 H), 4.36-4.19 (m, 1 H), 4.03-3.88 (m, 1 H), 3.78 (s, 3H), 2.08 (s, 3H).
Beispiel 56
(+/-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(2-fluor-6-methoxyphenyl)- propansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 2-(2-fluor-6-methoxyphenyl)propanoat (40 mg, 67 μηηοΙ, Beispiel 173A) in Dichlormethan (2 ml) wurde TFA (420 μΙ, 5.4 mmol) gegeben, und das Gemisch wurde 5 Tage bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlorme- than versetzt und wieder eingeengt. Der Rückstand wurde in Dichlormethan aufgenommen und mittels präparativer HPLC (Methode 15) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde in Aceton itril/Wasser lyophilisiert. Es wurden 34 mg (100% Reinheit, 93% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.85 min; MS (ESIpos): m/z = 537/539 [M+H]+
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: 1.234 (0.96), 2.173 (16.00), 3.754 (0.79), 4.098 (1.28), 4.109 (2.39), 4.121 (1.80), 4.135 (2.09), 4.147 (1.18), 4.356 (2.32), 4.367 (2.60), 4.373 (2.60), 4.385 (1.98), 6.804 (2.49), 6.822 (4.41 ), 6.840 (2.56), 6.878 (4.65), 6.895 (4.96), 7.273 (1.25), 7.289 (2.73), 7.303 (2.62), 7.320 (1.09), 7.474 (0.53), 7.477 (0.66), 7.482 (1.13), 7.491 (2.50), 7.493 (2.21 ), 7.497 (2.17), 7.503 (4.94), 7.505 (4.96), 7.508 (5.85), 7.522 (12.63), 7.528 (14.25), 7.532 (14.52), 7.544 (2.43), 7.753 (0.65), 7.843 (3.64), 7.847 (3.30), 7.860 (5.06), 7.865 (4.82), 7.932 (9.02), 7.949 (6.02), 8.785 (2.08), 8.797 (3.93), 8.809 (2.00).
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: 12.51 (br. s, 1 H), 8.80 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.75 (br. s, 1 H), 7.57-7.47 (m, 5H), 7.34-7.25 (m, 1 H), 6.89 (d, 1 H), 6.82 (t, 1 H), 4.37 (dd, 1 H), 4.19-4.06 (m, 1 H), 3.84-3.73 (m, 1 H, teilweise verdeckt), 3.80 (s, 3H), 2.17 (s, 3H). Beispiel 57
(-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(2-fluor-6-methoxyphenyl)- propansäure (Enantiomer 1)
Zu einer Lösung aus (-)-ie/f-Butyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2- (2-fluor-6-methoxyphenyl)propanoat (60 mg, 101 μηηοΙ, Beispiel 174A) in Dichlormethan (3 ml) wurde TFA (620 μΙ, 8.1 mmol) gegeben, und das Gemisch wurde einen Tag bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde in Dichlormethan aufgenommen und mittels präparativer HPLC (Methode 15) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde in Aceton itril/Wasser lyophilisiert. Es wurden 55 mg (100% Reinheit, ee-Wert >99%, 100% d. Th.) der Titelverbindung erhalten.
[a]D 20 = -82.2°, 589 nm, c = 0.42 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.86 min; MS (ESIpos): m/z = 537/539 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 2.171 (16.00), 3.743 (0.65), 3.764 (1.01 ), 4.088 (1.12), 4.102 (2.09), 4.1 17 (1.58), 4.136 (1.82), 4.150 (1.08), 4.348 (2.38), 4.363 (2.62), 4.370 (2.64),
4.385 (2.12), 6.799 (1.98), 6.821 (3.50), 6.843 (2.15), 6.876 (3.79), 6.897 (4.17), 7.267 (1.10), 7.288 (2.42), 7.306 (2.34), 7.326 (0.97), 7.479 (0.56), 7.493 (1.54), 7.503 (4.89), 7.51 1 (4.31 ), 7.523 (14.02), 7.526 (13.66), 7.535 (7.39), 7.744 (0.75), 7.842 (2.43), 7.846 (2.19), 7.864 (3.90), 7.869 (3.73), 7.930 (6.87), 7.952 (4.18), 8.781 (1.59), 8.796 (3.17), 8.811 (1.58).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.50 (br. s, 1 H), 8.80 (t, 1 H), 7.94 (d, 1 H), 7.86 (dd, 1 H), 7.74 (br. s, 1 H), 7.56-7.46 (m, 5H), 7.35-7.25 (m, 1 H), 6.89 (d, 1 H), 6.82 (t, 1 H), 4.37 (dd, 1 H), 4.17-4.06 (m, 1 H), 3.84-3.73 (m, 1 H, teilweise verdeckt), 3.80 (s, 3H), 2.17 (s, 3H).
Beispiel 58
(+)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(2-fl^
propansäure (Enantiomer 2)
Zu einer Lösung aus (+)-ie/f-Butyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2- (2-fluor-6-methoxyphenyl)propanoat (70 mg, 1 18 μηηοΙ, Beispiel 175A) in Dichlormethan (3.5 ml) wurde TFA (730 μΙ, 9.4 mmol) gegeben, und das Gemisch wurde einen Tag bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlor- methan versetzt und wieder eingeengt. Der Rückstand wurde in Dichlormethan aufgenommen und mittels praparativer HPLC (Methode 15) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde in Acetonitril/Wasser lyophilisiert. Es wurden zunächst 65 mg („100% Reinheit", ee-Wert > 99%,„>100% d. Th.", lösungsmittelhaltig) der Titelverbindung erhalten (s. Drehwert).
[a]D 20 = +80.0°, 589 nm, c = 0.40 g/100 ml, Methanol
Das lösungsmittelhaltige Produkt wurde erneut in Acetonitril/Wasser lyophilisiert (s. u. Analytik). Es wurden 55 mg (100 % Reinheit, 87 % d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 0.99 min; MS (ESIpos): m/z = 537/539 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.149 (0.69), -0.008 (7.09), 0.008 (5.65), 0.146 (0.72), 2.169 (16.00), 2.327 (1.61 ), 2.366 (1.38), 2.523 (5.32), 2.669 (1.64), 2.710 (1.35), 4.086 (1.44), 4.100 (2.33), 4.1 15 (1.73), 4.133 (1.97), 4.147 (1.20), 4.347 (1.94), 4.362 (2.18), 4.369 (2.15), 4.383 (1.61 ), 6.798 (2.06), 6.820 (3.56), 6.842 (2.21 ), 6.875 (3.83), 6.896 (4.19), 7.267 (1.14), 7.288 (2.39), 7.305 (2.39), 7.326 (0.99), 7.492 (1.61 ), 7.502 (5.02), 7.510 (4.52), 7.522 (14.71 ), 7.534 (7.06), 7.741 (0.81 ), 7.839 (2.60), 7.845 (2.27), 7.862 (4.10), 7.867 (3.83), 7.928 (7.33), 7.951 (4.46), 8.778 (1.59), 8.793 (3.14), 8.807 (1.59).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.52 (br. s, 1 H), 8.79 (t, 1 H), 7.94 (d, 1 H), 7.86 (dd, 1 H), 7.74 (br. s, 1 H), 7.56-7.46 (m, 5H), 7.35-7.24 (m, 1 H), 6.89 (d, 1 H), 6.82 (t, 1 H), 4.37 (dd, 1 H), 4.18-4.08 (m, 1 H), 3.80 (s, 3H), ca. 3.8 (1 H, verdeckt), 2.17 (s, 3H).
Beispiel 59
(■)_3_{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amin
propansäure (Enantiomer 1)
Zu einer Lösung aus (-)-ie/f-Butyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2- (2-methoxyphenyl)-2-methylpropanoat (400 mg, 678 μηηοΙ, Beispiel 177A) in Dichlormethan (10 ml) wurde TFA (1.0 ml, 14 mmol) gegeben, und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 303 mg (98% Reinheit, ee-Wert 99%, 82% d. Th.) der Titelverbindung erhalten.
[a]D 20 = -85.8°, 589 nm, c = 0.30 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.01 min; MS (ESIpos): m/z = 533/535 [M+H]+
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: -0.007 (0.46), 1.594 (16.00), 2.071 (2.90), 2.085 (1.42), 3.718 (1.59), 3.728 (1.74), 3.744 (1.87), 3.755 (1.76), 4.314 (1.91 ), 4.329 (2.1 1 ), 4.341 (1.94), 4.356 (1.79), 6.930 (1.35), 6.945 (2.81 ), 6.960 (1.62), 6.999 (2.72), 7.015 (3.20), 7.251 (5.49), 7.266 (6.00), 7.280 (1.18), 7.480 (0.72), 7.489 (1.76), 7.491 (1.53), 7.493 (1.43), 7.501 (4.35), 7.506 (4.43), 7.518 (1 1.88), 7.523 (1 1.44), 7.528 (10.05), 7.539 (1.98), 7.831 (2.65), 7.836 (2.51 ), 7.849 (3.84), 7.853 (3.79), 7.919 (7.44), 7.936 (5.00), 8.417 (1.69), 8.428 (2.27), 8.432 (2.33), 8.443 (1.73).
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: 12.15 (br. s, 1 H), 8.43 (dd, 1 H), 7.93 (d, 1 H), 7.84 (dd, 1 H), 7.64 (br. s, 1 H), 7.56-7.45 (m, 5H), 7.31-7.22 (m, 2H), 7.01 (d, 1 H), 6.95 (t, 1 H), 4.34 (dd, 1 H), 3.78 (s, 3H), 3.74 (dd, 1 H), 2.08 (br. s, 3H), 1.59 (s, 3H).
Beispiel 60
(+)-3-{[(6-Brom-3-methyl-2-phenylcN^
propansäure (Enantiomer 2)
Zu einer Lösung aus (+)-ie f-Butyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2- (2-methoxyphenyl)-2-methylpropanoat (400 mg, 678 μmol, Beispiel 178A) in Dichlormethan (10 ml) wurde TFA (1.0 ml, 14 mmol) gegeben, und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 133 mg (98% Reinheit, ee-Wert 95%, 36% d. Th.) der Titelverbindung erhalten.
[a]D 20 = +76.7°, 589 nm, c = 0.30 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.01 min; MS (ESIpos): m/z = 533/535 [M+H]+
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: -0.007 (0.43), 1.595 (16.00), 2.071 (2.05), 2.086 (1.47), 3.720 (1.58), 3.730 (1.73), 3.746 (1.89), 3.757 (1.78), 4.315 (1.89), 4.330 (2.10), 4.342 (1.94), 4.357 (1.78), 6.931 (1.36), 6.946 (2.83), 6.961 (1.66), 6.999 (2.72), 7.015 (3.24), 7.252 (5.37), 7.267 (5.93), 7.281 (1.28), 7.481 (0.66), 7.490 (1.70), 7.492 (1.49), 7.494 (1.37), 7.502 (4.46), 7.507 (4.51 ), 7.519 (1 1.46), 7.524 (1 1.39), 7.529 (10.46), 7.540 (2.28), 7.593 (0.40), 7.641 (0.41 ), 7.833 (2.58), 7.837 (2.55), 7.851 (3.77), 7.855 (3.88), 7.920 (7.33), 7.938 (4.95), 8.419 (1.68), 8.430 (2.29), 8.434 (2.40), 8.444 (1.78).
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: 8.43 (dd, 1 H), 7.93 (d, 1 H), 7.84 (dd, 1 H), 7.64 (br. s, 1 H), 7.56-7.46 (m, 5H), 7.30-7.22 (m, 2H), 7.01 (d, 1 H), 6.95 (t, 1 H), 4.34 (dd, 1 H), 3.79 (s, 3H), 3.74 (dd, 1 H), 2.09 (br. s, 3H), 1.60 (s, 3H).
Beispiel 61
(+)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(2-chlor-6-fluorphenyl)-2- methylpropansäure (Enantiomer 1)
Zu einer Lösung aus (+)-ie/f-Butyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2- (2-chlor-6-fluorphenyl)-2-methylpropanoat (150 mg, 245 μηηοΙ, Beispiel 180A) in Dichlormethan (1.7 ml) wurde TFA (380 μΙ, 4.9 mmol) gegeben, und das Gemisch wurde 7 h bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan ver- setzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 20) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 96 mg (98% Reinheit, ee-Wert 99%, 69% d. Th.) der Titelverbindung erhalten.
[a]D 20 = +68.8°, 589 nm, c = 0.38 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.97 min; MS (ESIpos): m/z = 555/557 [M+H]+
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: -0.006 (0.85), 0.006 (0.57), 1.196 (0.41 ), 1.209 (0.41 ), 1.838 (8.67), 1.851 (8.44), 2.073 (1.43), 2.167 (1.73), 2.270 (0.45), 2.518 (0.69), 2.522 (0.48), 3.850 (1.19), 4.466 (1.35), 7.168 (1.13), 7.185 (1.55), 7.208 (1.27), 7.314 (1.99), 7.329 (3.87), 7.351 (1.75), 7.482 (1.18), 7.485 (1.27), 7.494 (3.79), 7.501 (3.73), 7.507 (6.74), 7.51 1 (8.03), 7.525 (12.08), 7.537 (16.00), 7.553 (3.51 ), 7.843 (3.37), 7.847 (3.18), 7.861 (4.71 ), 7.865 (4.52), 7.934 (9.34), 7.952 (6.28), 8.769 (2.37), 8.783 (3.82), 8.795 (2.25).
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: 12.85 (br. s, 1 H), 8.78 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.68 (br. s, 1 H), 7.57-7.47 (m, 5H), 7.41-7.28 (m, 2H), 7.23-7.14 (m, 1 H), 4.54-4.39 (m, 1 H), 3.91-3.79 (m, 1 H), 2.17 (br. s, 3H), 1.84 (d, 3H).
Beispiel 62
(-)-3-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-(2-chlor-6-fluorphenyl)-2- methylpropansäure (Enantiomer 2)
Zu einer Lösung aus (-)-ie/f-Butyl-3-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2- (2-chlor-6-fluorphenyl)-2-methylpropanoat (150 mg, 245 μηηοΙ, Beispiel 181A) in Dichlormethan (1.7 ml) wurde TFA (380 μΙ, 4.9 mmol) gegeben, und das Gemisch wurde 7 h bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 20) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 1 12 mg (98% Reinheit, ee-Wert 97%, 81% d. Th.) der Titelverbindung erhalten. [a]D 20 = -71.7°, 589 nm, c = 0.29 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.97 min; MS (ESIpos): m/z = 555/557 [M+H]+
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: -0.007 (0.55), 0.006 (0.41 ), 1.840 (8.88), 1.853 (8.69), 2.073 (1.64), 2.168 (1.72), 2.271 (0.47), 2.518 (0.46), 3.837 (1.10), 3.851 (1.13), 4.468 (1.32), 4.480 (1.27), 7.168 (1.13), 7.185 (1.52), 7.208 (1.24), 7.315 (1.97), 7.329 (3.96), 7.351 (1.73),
7.477 (0.77), 7.482 (1.15), 7.485 (1.17), 7.494 (3.97), 7.502 (3.56), 7.507 (6.91 ), 7.51 1 (8.58), 7.525 (12.45), 7.539 (16.00), 7.554 (3.71 ), 7.843 (3.54), 7.848 (3.35), 7.861 (4.95), 7.866 (4.81 ), 7.935 (10.19), 7.953 (6.90), 8.772 (2.46), 8.785 (3.96), 8.798 (2.37).
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: 12.87 (br. s, 1 H), 8.79 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.70 (br. s, 1 H), 7.59-7.46 (m, 5H), 7.42-7.27 (m, 2H), 7.24-7.12 (m, 1 H), 4.53-4.41 (m, 1 H), 3.91-3.80 (m, 1 H), 2.17 (br. s, 3H), 1.85 (d, 3H).
Beispiel 63
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-ph (Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (150 mg, 438 μηηοΙ, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (1.5 ml) wurden HATU (250 mg, 658 μηηοΙ) und DIPEA (230 μΙ, 1.3 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-4-Amino-3-phenylbutansäure-Hydrochlorid (142 mg, 658 μηηοΙ, CAS-RN 3060-41-1 , kommerziell verfügbar), gelöst in DMF (1 ml) hinzugegeben, und das Gemisch wurde über Nacht bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch mittels präparativer HPLC (Methode 22) gereinigt. Es wurden 206 mg (96% Reinheit, 90% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.85 min; MS (ESIpos): m/z = 503/505 (M+H)+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 2.065 (1.35), 2.073 (6.33), 2.085 (1.59), 2.573 (1.56), 2.590 (2.18), 2.613 (2.18), 2.724 (2.06), 2.738 (2.28), 2.763 (1.51 ), 2.778 (1.45), 3.390 (0.91 ), 3.399 (1.09), 3.405 (0.95), 3.413 (1.61 ), 3.428 (1.05), 3.436 (0.89), 3.573 (0.84), 3.593 (0.91 ), 3.607 (1.10), 3.619 (0.64), 3.752 (0.68), 3.770 (0.95), 3.791 (0.87), 3.804 (0.73), 7.224 (1.76), 7.241 (1.64), 7.260 (0.79), 7.278 (1.07), 7.298 (2.13), 7.318 (6.13), 7.337 (12.61 ), 7.353 (1.88), 7.358 (1.04), 7.490 (1.44), 7.501 (4.52), 7.508 (4.98), 7.519 (16.00), 7.526 (7.90), 7.530 (5.64), 7.534 (4.95), 7.541 (1.29), 7.835 (2.04), 7.840 (1.87), 7.857 (3.22), 7.862 (2.99), 7.929 (6.06), 7.951 (3.77), 8.803 (1.31 ), 8.820 (1.87), 8.832 (1.28).
Trennunq der Enantiomere:
Die Titelverbindung (200 mg) wurde mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 64 und 65) [Säule: Daicel Chiralpak AD-H, 5 μηη, 250 mm x 20 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 1 ml; Eluent: 70% Koh- lendioxid / 30% Isopropanol; Laufzeit 70 min, isokratisch].
Beispiel 64
(-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-phenylbutansäure (Enantiomer 1)
Bei der in Beispiel 63 beschriebenen Enantiomerentrennung wurden 73 mg (98% Reinheit, ee-Wert 100%) der Titelverbindung als früher eluierendes Enantiomer erhalten. [a]D 20 = -26.3°, 589 nm, c = 0.32 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.87 min; MS (ESIpos): m/z = 503/505 (M+H)+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.76), 0.008 (0.75), 2.084 (1.66), 2.573 (1.59), 2.590 (2.19), 2.613 (2.18), 2.724 (2.14), 2.730 (1.00), 2.738 (2.33), 2.764 (1.55), 2.778 (1.45), 3.392 (0.98), 3.399 (1.13), 3.406 (1.03), 3.414 (1.66), 3.429 (1.09), 3.437 (0.91 ), 3.561 (0.61 ), 3.573 (0.90), 3.594 (0.99), 3.607 (1.16), 3.620 (0.66), 3.753 (0.71 ), 3.772 (1.02), 3.792 (0.90), 3.803 (0.77), 7.208 (1.1 1 ), 7.224 (1.97), 7.241 (1.77), 7.260 (0.84), 7.279 (1.22), 7.298 (2.49), 7.318 (6.59), 7.334 (13.17), 7.353 (1.90), 7.359 (1.00), 7.477 (0.79), 7.480 (0.87), 7.489 (1.74), 7.492 (1.94), 7.503 (5.07), 7.510 (5.62), 7.520 (16.00), 7.527 (7.93), 7.532 (5.62), 7.535 (4.55), 7.543 (1.04), 7.837 (2.19), 7.842 (1.93), 7.860 (3.35), 7.865 (2.92), 7.930 (5.95), 7.953 (3.66), 8.806 (1.46), 8.818 (1.95), 8.834 (1.25).
Beispiel 65
(+)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-phenylbutansäure (Enantiomer 2)
Bei der in Beispiel 63 beschriebenen Enantiomerentrennung wurde die vorgereinigte Titelverbin- dung als später eluierendes Enantiomer erhalten und mittels päparativer HPLC (Methode 21 ) nachgereinigt. Es wurden 65 mg (94% Reinheit, ee-Wert 100%) der Titelverbindung erhalten.
[a]D 20 = +26.4°, 589 nm, c = 0.35 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 0.98 min; MS (ESIpos): m/z = 503/505 (M+H)+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 2.083 (1.85), 2.572 (1.51 ), 2.723 (2.01 ), 2.738 (2.17), 2.763 (1.41 ), 2.777 (1.34), 3.391 (0.92), 3.399 (1 .09), 3.413 (1.64), 3.427 (1.16), 3.436 (0.96), 3.574 (0.97), 3.593 (1.09), 3.606 (1.27), 3.752 (0.72), 3.770 (1.09), 3.791 (1.03), 7.224 (1.64), 7.241 (1.57), 7.298 (1.72), 7.318 (5.68), 7.336 (13.29), 7.352 (2.78), 7.491 (1.18), 7.503 (3.89),
7.510 (4.60), 7.521 (16.00), 7.837 (1.86), 7.842 (1.88), 7.860 (3.04), 7.865 (3.10), 7.930 (5.1 1 ), 7.953 (3.46), 8.806 (1.26), 8.822 (2.18), 8.834 (1.62).
Beispiel 66
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]ami
(Racemat)
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (150 mg, 438 μηηοΙ, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (1.5 ml) wurden HATU (250 mg, 658 μηηοΙ) und DIPEA (230 μΙ, 1.3 mmol) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde (+/-)-4-Amino-3-(4-chlorphenyl)butansäure (140 mg, 658 μηηοΙ, CAS-RN 1 134-47-0, kommerziell verfügbar), gelöst in DMF (2 ml) hinzugegeben, und das Gemisch wurde 3 h bei 60 °C gerührt. Nach Abkühlen auf RT wurde das Gemisch in eine Zitronensäure-Lösung (50 ml) gegeben und geschüttelt. Der gebildete Niederschlag wurde abfiltriert, zweimal mit jeweils 5 ml Wasser gewaschen und mittels präparativer HPLC (Methode 15) gereinigt. Es wurden 97 mg (100% Reinheit, 41 % d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.09 min; MS (ESIpos): m/z = 537/539 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 2.086 (5.51 ), 2.568 (0.68), 2.608 (0.91 ), 2.725 (0.88), 2.738 (0.98), 2.764 (0.66), 7.380 (16.00), 7.505 (2.07), 7.513 (1.89), 7.524 (5.04), 7.529 (4.78), 7.532 (4.20), 7.536 (3.04), 7.540 (2.40), 7.836 (0.74), 7.841 (0.74), 7.859 (1.17), 7.864 (1.18), 7.932 (2.34), 7.955 (1.50), 8.826 (0.91 ).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 12.03 (br. s, 1 H), 8.82 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.65-7.46 (m, 6H), 7.38 (s, 4H), 3.79 (br. s, 1 H), 3.66-3.52 (m, 1 H), 3.47-3.34 (m, 1 H), 2.75 (dd, 1 H), 2.63-2.55 (m, 1 H, teilweise verdeckt), 2.20-2.02 (br. m, 3H).
Trennunq der Enantiomere:
Die Titelverbindung (70 mg) wurde in einem Gemisch aus Ethanol (2 ml) und Isohexan (3 ml) gelöst und mittels präparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 67 und 68) [Säule: Daicel Chiralpak ID, 5 μηη, 250 mm x 20 mm; Fluss: 15 ml/min; Tempera- tur: 30 °C; Injektion: 0.25 ml; Eluent: 82% Isohexan / 15% Ethanol + 0.2% Essigsäure; Laufzeit 22.5 min, isokratisch].
Beispiel 67
(+)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(4-chlorphenyl)butansäure (Enantiomer 1)
Bei der in Beispiel 66 beschriebenen Enantiomerentrennung wurde die vorgereinigte Titelverbindung als früher eluierendes Enantiomer erhalten und mittels päparativer HPLC (Methode 15) nachgereinigt. Es wurden 28 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung erhalten.
[a]D 20 = +40.9°, 589 nm, c = 0.33 g/100 ml, Chloroform
LC-MS (Methode 1 ): Rt = 1.95 min; MS (ESIpos): m/z = 537/539 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 2.583 (0.89), 2.606 (0.88), 2.723 (0.84), 2.737 (0.93), 7.378 (16.00), 7.504 (1.93), 7.512 (1.74), 7.523 (4.86), 7.528 (4.43), 7.532 (3.55), 7.536 (2.58), 7.540 (2.07), 7.836 (0.71 ), 7.858 (1.10), 7.863 (1.07), 7.931 (2.34), 7.954 (1.48).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 12.17 (br. s, 1 H), 8.82 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.65-7.46 (m, 6H), 7.38 (s, 4H), 3.79 (br. s, 1 H), 3.67-3.52 (m, 1 H), 3.47-3.32 (m, 1 H), 2.75 (dd, 1 H), 2.63-2.54 (m, 1 H), 2.12 (br. s, 3H).
Beispiel 68
(-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(4-chlorphenyl)butansäure (Enantiomer 2)
Bei der in Beispiel 66 beschriebenen Enantiomerentrennung wurde die vorgereinigte Titelverbin- dung als später eluierendes Enantiomer erhalten und mittels päparativer HPLC (Methode 15) nachgereinigt. Es wurden 30 mg (100% Reinheit, ee-Wert 94%) der Titelverbindung erhalten.
[a]D 20 = -39.8°, 589 nm, c = 0.33 g/100 ml, Chloroform
LC-MS (Methode 1 ): Rt = 1.95 min; MS (ESIpos): m/z = 537/539 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 2.583 (0.90), 2.607 (0.87), 2.723 (0.84), 2.738 (0.93), 7.379 (16.00), 7.505 (2.05), 7.513 (1.93), 7.524 (5.10), 7.529 (4.75), 7.532 (3.91 ), 7.536 (2.87), 7.540 (2.25), 7.837 (0.77), 7.842 (0.73), 7.859 (1.19), 7.864 (1.14), 7.932 (2.42), 7.955 (1.51 ), 8.825 (0.85).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.82 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.69-7.43 (m, 6H), 7.38 (s, 4H), 3.80 (br. s, 1 H), 3.65-3.53 (m, 1 H), 3.48-3.35 (m, 1 H), 2.75 (dd, 1 H), 2.63-2.55 (m, 1 H), 2.1 1 (br. s, 3H).
Beispiel 69
(+/-)-4-{[(6-Brom-3-methyl-2-phenylcN^
Zu einer Lösung aus (+/-)-ierf-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 3-(2-chlorphenyl)butanoat (527 mg, 887 μηηοΙ, Beispiel 182A) in Dichlormethan (14 ml) wurde TFA (6.8 ml, 89 mmol) gegeben und das Gemisch wurde 2 h bei RT gerührt. Anschließend wur- de das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 22) gereinigt. Es wurden 340 mg (98% Reinheit, 70% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.02 min; MS (ESIpos): m/z = 537/539 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.150 (0.40), -0.008 (3.25), 0.008 (3.06), 2.073 (8.1 1 ), 2.142 (8.84), 2.192 (0.44), 2.327 (0.65), 2.332 (0.49), 2.523 (1.85), 2.645 (1.30), 2.664 (1.84), 2.674 (0.80), 2.685 (3.47), 2.705 (3.43), 2.723 (3.28), 2.739 (3.51 ), 2.763 (1.31 ), 2.779 (1.23), 3.615 (2.15), 3.803 (1.09), 3.932 (1.69), 3.951 (2.09), 3.969 (1.30), 7.246 (1.28), 7.265 (2.98), 7.281 (2.15), 7.334 (1.92), 7.353 (3.15), 7.371 (1.55), 7.428 (4.71 ), 7.431 (4.62), 7.447 (3.77), 7.451 (3.63), 7.476 (1.01 ), 7.487 (2.17), 7.501 (6.90), 7.509 (8.95), 7.512 (6.82), 7.521 (14.80), 7.527 (16.00), 7.530 (14.59), 7.534 (10.78), 7.538 (7.36), 7.546 (2.23), 7.648 (0.94), 7.833 (3.21 ), 7.838 (2.95), 7.855 (4.93), 7.860 (4.75), 7.929 (9.28), 7.952 (5.83), 8.832 (1.79), 8.847 (3.52), 8.862 (1.73).
Trennung der Enantiomere:
Die Titelverbindung (200 mg) wurde in Acetonitril (18 ml) gelöst und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 70 und 71 ) [Säule: Daicel Chiralpak
AD, 5 μηι, 250 mm x 20 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 0.3 ml; Eluent: 80% Kohlendioxid / 20% Methanol; Laufzeit 12 min, isokratisch].
Beispiel 70
(+)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-chlorphenyl)butansäure (Enantiomer 1)
Bei der in Beispiel 69 beschriebenen Enantiomerentrennung wurden 44 mg (100% Reinheit, ee-Wert 100%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = +16.8°, 589 nm, c = 0.40 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.92 min; MS (ESIpos): m/z = 537/539 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 2.143 (8.97), 2.327 (0.46), 2.631 (0.98), 2.649 (1.09), 2.670 (3.05), 2.689 (2.67), 2.709 (2.71 ), 2.725 (2.59), 2.749 (1.00), 2.765 (0.95), 3.31 1 (1.98), 3.71 1 (1.24), 3.813 (0.94), 3.929 (1.51 ), 3.945 (1.95), 3.963 (1.23), 7.243 (1.30), 7.261 (3.01 ), 7.280 (2.15), 7.332 (1.87), 7.350 (3.13), 7.368 (1.51 ), 7.427 (4.71 ), 7.447 (3.84), 7.475 (0.85), 7.501 (7.67), 7.508 (8.77), 7.520 (14.62), 7.527 (16.00), 7.656 (1.05), 7.831 (3.10), 7.836 (2.78), 7.853 (4.62), 7.858 (4.42), 7.928 (8.78), 7.950 (5.48), 8.875 (2.67).
Beispiel 71
(-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-chlorphenyl)butansäure (Enantiomer 2)
Bei der in Beispiel 69 beschriebenen Enantiomerentrennung wurden 61 mg (98% Reinheit, ee-Wert 98%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = -18.1 °, 589 nm, c = 0.37 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.92 min; MS (ESIpos): m/z = 537/539 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.284 (2.85), 2.144 (9.37), 2.193 (0.50), 2.523 (0.75), 2.646 (1.24), 2.665 (1.48), 2.686 (3.35), 2.705 (3.37), 2.723 (3.22), 2.740 (3.43), 2.764 (1.30), 2.780 (1.21 ), 3.536 (1.08), 3.718 (1.28), 3.815 (0.99), 3.917 (0.65), 3.935 (1.69), 3.953 (2.12), 3.970 (1.36), 3.988 (0.46), 7.246 (1.35), 7.264 (3.13), 7.282 (2.27), 7.334 (2.01 ), 7.353 (3.31 ), 7.371 (1.64), 7.429 (4.72), 7.431 (4.62), 7.449 (3.85), 7.475 (0.94), 7.487 (2.24), 7.501 (7.16), 7.508 (8.46), 7.513 (7.20), 7.520 (15.06), 7.527 (16.00), 7.529 (15.63), 7.546 (2.45), 7.663 (0.98), 7.832 (3.10), 7.837 (2.83), 7.854 (4.76), 7.859 (4.58), 7.929 (8.85), 7.952 (5.65), 8.835 (1.83), 8.849 (3.57), 8.864 (1.82), 12.186 (1.34).
Beispiel 72
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amin
(Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 3-(3-chlorphenyl)butanoat (220 mg, 370 μηηοΙ, Beispiel 183A) in Dichlormethan (6.0 ml) wurde TFA (2.9 ml, 37 mmol) gegeben und das Gemisch wurde 2 h bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 22) gereinigt. Es wurden 93 mg (96% Reinheit, 45% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.95 min; MS (ESIpos): m/z = 537/539 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.62), 0.008 (0.73), 1.513 (4.18), 2.073 (1.21 ), 2.592 (1.53), 2.615 (1.62), 2.632 (2.44), 2.655 (2.46), 2.739 (2.29), 2.753 (2.58), 2.779 (1.63), 2.793 (1.51 ), 3.381 (0.92), 3.390 (1.07), 3.404 (1.69), 3.418 (1.18), 3.427 (1.00), 3.570 (0.82), 3.582 (1.22), 3.595 (0.96), 3.603 (1.23), 3.615 (1.57), 3.628 (0.98), 3.781 (1.47), 3.799 (1.84), 3.805 (1.82), 3.814 (1.83), 3.822 (1.95), 3.832 (1.89), 3.838 (1.86), 3.855 (1.78), 7.280 (1.27), 7.298 (2.12), 7.306 (1.72), 7.31 1 (2.01 ), 7.315 (1.69), 7.326 (3.60), 7.330 (5.17), 7.334 (3.33), 7.339 (4.65), 7.357 (3.12), 7.376 (1.02), 7.408 (5.29), 7.412 (3.39), 7.486 (0.77), 7.489 (1.30), 7.492 (1.48), 7.497 (1.47), 7.502 (4.59), 7.510 (4.68), 7.521 (16.00), 7.528 (7.86), 7.532 (5.84), 7.536 (5.32), 7.543 (1.39), 7.836 (1.84), 7.841 (1.74), 7.858 (2.85), 7.864 (2.81 ), 7.930 (6.1 1 ), 7.952 (3.87), 8.805 (1.47), 8.816 (1.89), 8.822 (1.94), 8.833 (1.42).
Trennung der Enantiomere:
Die Titelverbindung (220 mg) wurde in Ethanol (2 ml) gelöst und mittels präparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 73 und 74) [Säule: Daicel Chiralpak AZ-H, 5 μη-ι, 250 mm x 20 mm; Fluss: 20 ml/min; Detektion: 220 nm; Temperatur: 23 °C; Injektion: 0.05 ml; Eluent: 70% Heptan / 30% Ethanol + 0.2% TFA; isokratisch].
Beispiel 73
4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]^ (Enan- tiomer 1 )
Bei der in Beispiel 72 beschriebenen Enantiomerentrennung wurden 8 mg (90% Reinheit, ee-Wert 81 %) der Titelverbindung als früher eluierendes Enantiomer erhalten.
LC-MS (Methode 1 ): Rt = 1.93 min; MS (ESIpos): m/z = 537/539 [M+H]+
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: 0.844 (0.61 ), 0.858 (0.64), 1.090 (0.69), 1.104 (1.06), 1.1 18 (0.89), 1.148 (3.99), 1.163 (6.78), 1.177 (3.40), 1.233 (1.25), 1.257 (0.74), 2.067 (1.52), 2.600 (1.45), 2.618 (1.68), 2.632 (2.25), 2.650 (2.00), 2.745 (2.35), 2.756 (2.36), 2.777 (1.59), 2.788 (1.38), 2.906 (1.53), 2.920 (2.35), 2.931 (2.19), 2.945 (1.21 ), 3.407 (2.39), 3.587 (2.01 ), 3.612 (2.20), 3.730 (0.92), 3.744 (0.92), 3.790 (1.71 ), 3.804 (2.25), 3.848 (1.30), 7.298 (2.88), 7.316 (2.92), 7.330 (5.58), 7.341 (4.20), 7.356 (3.37), 7.371 (1.43), 7.408 (6.05), 7.509 (6.59), 7.523 (16.00), 7.842 (2.44), 7.859 (3.28), 7.933 (4.45), 7.951 (3.14), 8.823 (2.93).
Beispiel 74
4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(3-chlorphenyl)butansäure (Enantiomer 2)
Bei der in Beispiel 72 beschriebenen Enantiomerentrennung wurden 8 mg (90% Reinheit, ee-Wert 77%) der Titelverbindung als später eluierendes Enantiomer erhalten.
LC-MS (Methode 1 ): Rt = 1.93 min; MS (ESIpos): m/z = 537 [M+H]+
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: 0.845 (0.64), 0.859 (0.80), 1.090 (1.19), 1.104 (2.22), 1.1 18 (1.23), 1.148 (1.87), 1.162 (3.65), 1.177 (1.89), 1.233 (1.16), 1.246 (0.80), 1.258 (0.76), 2.073 (1.36), 2.602 (1.97), 2.620 (2.14), 2.634 (3.02), 2.652 (2.87), 2.746 (2.92), 2.757 (3.08), 2.778 (2.10), 2.790 (1.89), 2.925 (0.79), 2.935 (0.77), 3.380 (0.71 ), 3.391 (1.40), 3.398 (1.64), 3.409 (2.39), 3.420 (1.66), 3.427 (1.34), 3.439 (0.69), 3.581 (1.16), 3.590 (1.67), 3.601 (1.37), 3.607 (1.59), 3.617 (1.97), 3.626 (1.14), 3.730 (1.14), 3.744 (1.15), 3.792 (1.25), 3.807 (1.66), 3.819 (1.53), 3.825 (1.55), 3.833 (1.39), 3.852 (0.91 ), 7.284 (1.85), 7.299 (2.56), 7.316 (2.70), 7.331 (6.33), 7.342 (4.73), 7.357 (3.94), 7.373 (1.36), 7.381 (0.69), 7.410 (6.99), 7.489 (1.20), 7.498 (2.45), 7.510 (6.15), 7.515 (5.99), 7.526 (16.00), 7.530 (14.69), 7.536 (1 1.79), 7.546 (2.42), 7.849 (2.50), 7.853 (2.45), 7.867 (3.55), 7.871 (3.43), 7.940 (6.73), 7.957 (4.55), 8.817 (2.13), 8.826 (2.80), 8.831 (2.86), 8.840 (2.1 1 ).
Beispiel 75
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-methylphenyl)butansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 3-(2-methylphenyl)butanoat (750 mg, 1.31 mmol, Beispiel 184A) in Dichlormethan (21 ml) wurde TFA (10 ml, 130 mmol) gegeben und das Gemisch wurde 2 h bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 22) gereinigt. Es wurden 574 mg (98% Reinheit, 83% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.91 min; MS (ESIpos): m/z = 517/519 [M+H]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.195 (0.56), 1.21 1 (0.57), 2.072 (1.53), 2.1 18 (2.44), 2.398 (16.00), 2.580 (0.92), 2.602 (0.93), 2.620 (1.80), 2.641 (1.77), 2.701 (1.65), 2.715 (1.71 ), 2.740 (1.10), 2.754 (0.97), 3.475 (0.88), 3.491 (0.99), 3.504 (0.98), 3.714 (0.80), 3.729 (1.18), 3.755 (1.42), 3.771 (1.22), 3.785 (0.96), 3.803 (0.96), 7.087 (0.69), 7.105 (1.83), 7.122 (1.72), 7.151 (3.03), 7.171 (2.05), 7.193 (1.76), 7.210 (0.88), 7.348 (2.62), 7.366 (2.04), 7.490 (1 .38), 7.505 (4.13), 7.51 1 (3.27), 7.516 (2.22), 7.522 (7.34), 7.528 (4.36), 7.533 (8.00), 7.535 (7.96), 7.539 (5.03), 7.543 (3.79), 7.552 (1.44), 7.841 (1.92), 7.846 (1.73), 7.864 (2.88), 7.869 (2.73), 7.940 (5.41 ), 7.962 (3.45), 8.855 (1.14), 8.871 (1.89), 8.883 (1.04).
Trennung der Enantiomere:
Die Titelverbindung (750 mg) wurde mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 76 und 77) [Säule: Daicel Chiralpak AD-H, 5 μηη, 250 mm x 20 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 0.2 ml; Eluent: 85% Kohlendioxid / 15% Ethanol; isokratisch].
Beispiel 76
(-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-methylphenyl)butansäure (Enantiomer 1)
Bei der in Beispiel 75 beschriebenen Enantiomerentrennung wurde die Titelverbindung als früher eluierendes Enantiomer erhalten. Es wurden 188 mg (98% Reinheit, ee-Wert 100%) der Titelverbindung erhalten.
[a]D 20 = -23.3°, 589 nm, c = 0.29 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.08 min; MS (ESIpos): m/z = 517/519 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.53), 0.008 (0.42), 1.031 (1.36), 1.046 (1.36), 1.285 (0.75), 2.072 (0.86), 2.1 16 (2.61 ), 2.396 (16.00), 2.522 (0.65), 2.579 (0.93), 2.600 (0.96), 2.618 (1.83), 2.640 (1.77), 2.700 (1.69), 2.713 (1.75), 2.739 (1.12), 2.753 (0.98), 3.471 (0.96), 3.487 (1.07), 3.502 (1.06), 3.516 (0.67), 3.712 (0.84), 3.727 (1.25), 3.753 (1.51 ), 3.769 (1.30), 3.783 (1.01 ), 3.800 (0.98), 3.820 (0.46), 7.085 (0.72), 7.104 (1.90), 7.122 (1.82), 7.150 (3.13), 7.170 (2.15), 7.192 (1.82), 7.210 (0.89), 7.347 (2.72), 7.366 (2.08), 7.475 (0.58), 7.488 (1.59), 7.502 (4.47), 7.508 (3.75), 7.520 (7.82), 7.530 (8.17), 7.532 (8.13), 7.548 (1.47), 7.675 (0.67), 7.837 (2.01 ), 7.842 (1.81 ), 7.859 (2.92), 7.864 (2.77), 7.936 (5.33), 7.959 (3.38), 8.849 (1.23), 8.865 (1.95), 8.877 (1.07), 12.073 (1.54). Beispiel 77
(+)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-methylphenyl)butansäure (Enantiomer 2)
Bei der in Beispiel 75 beschriebenen Enantiomerentrennung wurde die Titelverbindung als später eluierendes Enantiomer erhalten. Es wurden 179 mg (98% Reinheit, ee-Wert 97%) der Titel- Verbindung erhalten.
[a]D 20 = +24.8°, 589 nm, c = 0.38 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.08 min; MS (ESIpos): m/z = 517/519 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.42), 1.030 (1.22), 1.046 (1.23), 1.284 (0.59), 2.073 (0.85), 2.1 16 (2.44), 2.396 (16.00), 2.523 (0.47), 2.578 (0.91 ), 2.599 (0.93), 2.617 (1.82), 2.639 (1.78), 2.699 (1.65), 2.712 (1.74), 2.738 (1.1 1 ), 2.752 (0.99), 3.471 (0.90), 3.487 (1.01 ),
3.501 (1.01 ), 3.516 (0.66), 3.71 1 (0.78), 3.726 (1.17), 3.752 (1.41 ), 3.768 (1.22), 3.782 (0.97), 3.800 (0.96), 3.819 (0.46), 7.086 (0.66), 7.104 (1.82), 7.122 (1.73), 7.150 (2.98), 7.170 (2.01 ), 7.192 (1.74), 7.210 (0.87), 7.346 (2.67), 7.365 (2.07), 7.475 (0.48), 7.488 (1.38), 7.493 (1.00),
7.502 (4.10), 7.509 (3.35), 7.521 (7.50), 7.529 (7.78), 7.532 (7.90), 7.536 (5.27), 7.548 (1.38), 7.674 (0.60), 7.837 (1.93), 7.843 (1.74), 7.860 (2.90), 7.865 (2.79), 7.936 (5.20), 7.959 (3.34),
8.849 (1.15), 8.865 (1.90), 8.877 (1.05), 12.073 (2.06).
Beispiel 78
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amin
butansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 3-(2,6-dichlorphenyl)butanoat (140 mg, 223 μmol, Beispiel 185A) in Dichlormethan (3.6 ml) wurde TFA (1.7 ml, 22 mmol) gegeben und das Gemisch wurde 2 h bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 22) gereinigt. Es wurden 103 mg (97% Reinheit, 78% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.96 min; MS (ESIpos): m/z = 571/573/575 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.34), 0.008 (1.24), 1.194 (0.98), 1.21 1 (0.93), 1.491 (1.86), 2.073 (1.44), 2.170 (7.32), 2.881 (1.47), 2.898 (1.68), 2.921 (4.94), 2.937 (4.87), 2.948 (4.80), 2.968 (4.74), 2.987 (1.53), 3.008 (1.61 ), 3.628 (1.72), 3.801 (1.46), 4.1 18 (1.23), 4.414 (1.41 ), 4.430 (1.84), 4.450 (1.22), 7.266 (1.46), 7.286 (2.89), 7.307 (1.90), 7.424 (3.69), 7.444 (3.01 ), 7.464 (4.75), 7.488 (5.02), 7.495 (2.42), 7.504 (8.15), 7.51 1 (7.06), 7.515 (4.09), 7.523 (15.90), 7.530 (16.00), 7.533 (14.69), 7.537 (10.88), 7.541 (8.23), 7.550 (2.61 ), 7.838 (4.04), 7.843 (3.56), 7.860 (6.03), 7.866 (5.58), 7.933 (10.89), 7.956 (6.77), 8.914 (1.97), 8.929 (3.71 ), 8.944 (1.94).
Trennung der Enantiomere:
Die Titelverbindung (80 mg) wurde in 17 ml eines Methanol-Acetonitril-Gemisches gelöst und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 79 und 80) [Säule: Daicel Chiralpak AD-H, 5 μηι, 250 mm x 20 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 3 ml; Eluent: 70% Kohlendioxid / 30% Isopropanol; isokra- tisch].
Beispiel 79
4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2,6-di
(Enantiomer 1)
Bei der in Beispiel 78 beschriebenen Enantiomerentrennung wurde die Titelverbindung als früher eluierendes Enantiomer erhalten und mittels paparativer HPLC (Methode 21 ) nachgereinigt. Es wurden 22 mg (98% Reinheit, ee-Wert 96%) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.04 min; MS (ESIpos): m/z = 571/573/575 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 2.073 (0.63), 2.168 (7.87), 2.314 (0.56), 2.327 (0.72), 2.366 (0.53), 2.670 (0.66), 2.710 (0.43), 2.879 (1.34), 2.896 (1.55), 2.919 (4.81 ), 2.936 (4.87), 2.945 (4.79), 2.966 (4.59), 2.985 (1.43), 3.006 (1.44), 3.814 (1.32), 4.1 19 (1.35), 4.409 (1.91 ), 4.428 (2.43), 4.446 (1.78), 4.716 (0.73), 7.267 (1.34), 7.287 (2.87), 7.307 (1.97), 7.424 (3.51 ), 7.444 (2.89), 7.465 (4.64), 7.485 (4.08), 7.505 (7.47), 7.511 (6.25), 7.524 (16.00), 7.531 (15.93), 7.740 (0.43), 7.840 (3.42), 7.846 (3.16), 7.863 (5.30), 7.868 (5.12), 7.934 (9.44), 7.956 (5.90), 8.914 (2.03), 8.929 (3.94), 8.943 (2.10). Beispiel 80
4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2,6-dichlorphenyl)butansäure (Enantiomer 2)
Bei der in Beispiel 78 beschriebenen Enantiomerentrennung wurde die Titelverbindung als später eluierendes Enantiomer erhalten und mittels paparativer HPLC (Methode 21 ) nachgereinigt. Es wurden 30 mg (98% Reinheit, ee-Wert 95%) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.04 min; MS (ESIpos): m/z = 571/573/575 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.08), 0.008 (1.82), 2.168 (7.58), 2.327 (0.87), 2.366 (0.54), 2.669 (0.80), 2.710 (0.55), 2.879 (1.42), 2.896 (1.69), 2.919 (4.96), 2.936 (4.95), 2.945 (4.83), 2.966 (4.71 ), 2.985 (1.53), 3.006 (1.57), 3.814 (1.27), 4.1 18 (1.41 ), 4.41 1 (2.21 ), 4.428 (2.71 ), 7.267 (1.35), 7.286 (2.83), 7.307 (1.96), 7.424 (3.66), 7.444 (2.99), 7.464 (4.84), 7.484 (4.25), 7.504 (7.70), 7.51 1 (6.69), 7.524 (16.00), 7.530 (15.92), 7.549 (2.71 ), 7.723 (0.46), 7.839 (3.81 ), 7.845 (3.49), 7.862 (5.78), 7.867 (5.47), 7.933 (10.51 ), 7.956 (6.50), 8.913 (2.01 ), 8.928 (3.84), 8.943 (2.01 ).
Beispiel 81
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(3-methoxyphenyl)- butansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 3-(3-methoxyphenyl)butanoat (220 mg, 373 μηηοΙ, Beispiel 186A) in Dichlormethan (6.1 ml) wurde TFA (2.9 ml, 37 mmol) gegeben und das Gemisch wurde 2 h bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 22) gereinigt. Es wurden 145 mg (99% Reinheit, 72% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.83 min; MS (ESIpos): m/z = 533/535 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.517 (0.61 ), 2.073 (1.41 ), 2.100 (1.94), 2.566 (1.54), 2.583 (2.22), 2.606 (2.20), 2.706 (2.1 1 ), 2.721 (2.33), 2.746 (1.54), 2.760 (1.42), 3.365 (0.91 ), 3.372 (1.03), 3.387 (1.66), 3.402 (1.10), 3.409 (0.94), 3.558 (0.75), 3.571 (1.09), 3.585 (0.96), 3.591 (1.18), 3.604 (1.43), 3.618 (0.85), 3.746 (1.1 1 ), 3.762 (1.17), 3.778 (1.06), 3.784 (1.09), 3.795 (0.92), 3.817 (0.63), 6.782 (1.71 ), 6.800 (1.75), 6.805 (1.81 ), 6.895 (6.74), 6.899 (7.77), 6.917 (3.26), 7.212 (2.22), 7.231 (3.46), 7.251 (1.63), 7.490 (1.27), 7.492 (1.44), 7.503 (4.41 ), 7.51 1 (4.41 ), 7.522 (16.00), 7.529 (8.04), 7.533 (6.09), 7.536 (5.54), 7.544 (1.30), 7.835 (2.08), 7.840 (1.97), 7.857 (3.16), 7.863 (3.14), 7.931 (6.42), 7.954 (4.05), 8.795 (1.39), 8.81 1 (2.08), 8.823 (1.41 ).
Trennung der Enantiomere:
Die Titelverbindung (120 mg) wurde in 4 ml eines Ethanol-Heptan-Gemisches gelöst und mittels praparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 82 und 83) [Säule: Daicel Chiralpak AZ-H, 5 μηη, 250 mm x 20 mm; Fluss: 20 ml/min; Detektion: 220 nm; Temperatur: 40 °C; Injektion: 0.5 ml; Eluent: 70% Heptan / 30% Ethanol + 0.2% TFA; isokra- tisch, Laufzeit 8.5 min].
Beispiel 82
(+)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(3-methoxyphenyl)butansäure (Enantiomer 1)
Bei der in Beispiel 81 beschriebenen Enantiomerentrennung wurde die Titelverbindung als früher eluierendes Enantiomer erhalten. Es wurden 29 mg (98% Reinheit, ee-Wert 100%) der Titelverbindung erhalten.
[a]D 20 = +33.7°, 589 nm, c = 0.29 g/100 ml, Chloroform
LC-MS (Methode 1 ): Rt = 1.82 min; MS (ESIpos): m/z = 533/535 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.93), 0.008 (1.70), 1.038 (0.98), 1.056 (1.91 ), 1.073 (0.96), 1.207 (1.03), 1.234 (0.68), 2.073 (1.46), 2.101 (1.94), 2.523 (1.89), 2.564 (1.66), 2.581 (2.19), 2.604 (2.10), 2.705 (2.08), 2.719 (2.22), 2.744 (1.47), 2.759 (1.34), 3.362 (0.95), 3.369 (1.04), 3.384 (1.62), 3.400 (1.09), 3.407 (0.99), 3.414 (0.68), 3.432 (1.09), 3.449 (1.04), 3.556 (0.81 ), 3.568 (1.12), 3.589 (1.22), 3.602 (1.42), 3.615 (0.86), 3.743 (1.21 ), 3.759 (1.37), 3.782 (1.29), 3.793 (1.15), 3.816 (0.85), 3.996 (1.01 ), 6.780 (1.72), 6.804 (1.72), 6.894 (6.53), 6.898 (7.12), 6.915 (3.02), 7.210 (2.14), 7.230 (3.25), 7.250 (1.51 ), 7.489 (1.68), 7.502 (4.67), 7.510 (4.99), 7.521 (16.00), 7.528 (7.49), 7.532 (5.66), 7.535 (4.90), 7.834 (2.12), 7.839 (1.91 ), 7.856 (3.12), 7.862 (2.88), 7.930 (5.97), 7.952 (3.71 ), 8.792 (1.44), 8.809 (1.99), 8.821 (1.30). Beispiel 83
(-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(3-methoxyphenyl)butansäure (Enantiomer 2)
Bei der in Beispiel 81 beschriebenen Enantiomerentrennung wurde die Titelverbindung als später eluierendes Enantiomer erhalten. Es wurden 35 mg (98% Reinheit, ee-Wert 94%) der Titel- Verbindung erhalten.
[a]D 20 = -25.8°, 589 nm, c = 0.32 g/100 ml, Chloroform
LC-MS (Methode 1 ): Rt = 1.82 min; MS (ESIpos): m/z = 533/535 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.83), 0.008 (1.04), 0.840 (6.59), 0.858 (16.00), 0.875 (5.02), 1.242 (1 1.36), 1.247 (1 1.78), 1.258 (4.1 1 ), 1.267 (2.90), 1.279 (2.40), 1.297 (1.03), 2.097 (1.48), 2.564 (1.22), 2.581 (1.75), 2.604 (1.74), 2.705 (1.69), 2.719 (1.84), 2.744 (1.23), 2.759 (1.12), 3.363 (1.03), 3.370 (1.15), 3.385 (1.76), 3.399 (1.49), 3.407 (1.48), 3.422 (1.43), 3.432 (1.64), 3.450 (2.18), 3.467 (2.38), 3.535 (1.05), 3.544 (0.97), 3.556 (1.1 1 ), 3.569 (1.26), 3.583 (1.06), 3.589 (1.19), 3.602 (1.33), 3.759 (0.90), 6.781 (1.30), 6.804 (1.36), 6.894 (5.26), 6.898 (5.93), 6.916 (2.49), 7.210 (1.76), 7.230 (2.71 ), 7.250 (1.26), 7.488 (1.02), 7.491 (1.05), 7.501 (3.41 ), 7.509 (3.48), 7.520 (12.89), 7.527 (5.81 ), 7.532 (4.48), 7.535 (3.98), 7.832 (1.68), 7.838 (1.49), 7.855 (2.55), 7.860 (2.38), 7.929 (5.01 ), 7.952 (3.16), 8.792 (1.07), 8.808 (1.58), 8.820 (1.05).
Beispiel 84
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amin
methylbutansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 3-(3-chlorpyridin-2-yl)-3-methylbutanoat (650 mg, 1.07 mmol, Beispiel 187A) in Dichlormethan (7.2 ml) wurde TFA (1.6 ml, 21 mmol) gegeben und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash- Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohe- xan/Ethylacetat 8:2, Isolera One) gereinigt. Es wurden 202 mg (98% Reinheit, 34% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.94 min; MS (ESIpos): m/z = 552/554 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.685 (12.85), 2.073 (1.34), 2.163 (16.00), 2.518 (0.84), 2.523 (0.61 ), 2.762 (2.25), 2.803 (2.54), 3.285 (2.17), 3.326 (1.94), 4.000 (0.48), 4.016 (0.55), 4.034 (0.81 ), 4.049 (0.77), 4.110 (0.87), 4.124 (0.93), 4.143 (0.67), 4.158 (0.63), 4.347 (0.43), 7.287 (2.15), 7.298 (2.17), 7.307 (2.26), 7.318 (2.32), 7.490 (1.13), 7.505 (3.27), 7.51 1 (2.48), 7.517 (1.62), 7.522 (5.09), 7.530 (2.23), 7.536 (6.91 ), 7.545 (2.41 ), 7.552 (1.07), 7.555 (1.18), 7.707 (3.35), 7.712 (3.56), 7.830 (2.30), 7.834 (2.42), 7.839 (1.78), 7.844 (1.60), 7.850 (2.27), 7.853 (2.19), 7.861 (2.59), 7.867 (2.41 ), 7.933 (4.39), 7.955 (2.76), 8.459 (2.26), 8.463 (2.29), 8.471 (2.29), 8.474 (2.1 1 ), 8.678 (0.85), 8.693 (1.78), 8.709 (0.84).
Trennung der Enantiomere:
Die Titelverbindung (175 mg) wurde in 5 ml Isopropanol gelöst und mittels präparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 85 und 86) [Säule: YMC Chiralart Amylose SA, 5 μηη, 250 mm x 30 mm; Fluss: 30 ml/min; Detektion: 220 nm; Temperatur: 30 °C; Injektion: 0.35 ml; Eluent: 70% Heptan / 30% Isopropanol + 0.2% TFA; isokratisch, Laufzeit 14.5 min].
Beispiel 85
(-)-4-{[(6-Brom-3-methyl-2-phenylcN^
butansäure (Enantiomer 1)
Bei der in Beispiel 84 beschriebenen Enantiomerentrennung wurde die vorgereinigte Titelverbin- dung als früher eluierendes Enantiomer erhalten und mittels paparativer HPLC (Methode 20) nachgereinigt. Es wurden 69 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung erhalten.
[a]D 20 = -29.1 °, 589 nm, c = 0.31 g/100 ml, Chloroform
LC-MS (Methode 2): Rt = 1.02 min; MS (ESIpos): m/z = 552/554 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (0.71 ), 1.686 (13.36), 2.164 (16.00), 2.763 (2.29), 2.804 (2.60), 3.286 (2.29), 3.326 (2.03), 4.001 (0.42), 4.017 (0.49), 4.034 (0.77), 4.050 (0.72), 4.1 1 1 (0.77), 4.125 (0.81 ), 4.144 (0.50), 4.159 (0.44), 7.287 (1.99), 7.298 (2.04), 7.307 (2.1 1 ), 7.318 (2.13), 7.491 (1.19), 7.506 (3.51 ), 7.51 1 (2.65), 7.524 (5.56), 7.536 (7.08), 7.555 (1.24), 7.709 (3.45), 7.714 (3.66), 7.830 (2.18), 7.834 (2.30), 7.840 (1.74), 7.845 (1.64), 7.850 (2.20), 7.854 (2.1 1 ), 7.862 (2.53), 7.868 (2.35), 7.934 (4.34), 7.956 (2.71 ), 8.460 (2.14), 8.463 (2.18), 8.471 (2.15), 8.475 (2.02), 8.679 (0.93), 8.695 (1.93), 8.710 (0.89).
Beispiel 86
(+)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(3-chlorpyridin-2-yl)-3-methyl- butansäure (Enantiomer 2)
Bei der in Beispiel 84 beschriebenen Enantiomerentrennung wurde die vorgereinigte Titelverbin- dung als später eluierendes Enantiomer erhalten und mittels päparativer HPLC (Methode 20) nachgereinigt. Es wurden 73 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung erhalten.
[a]D 20 = +30.4°, 589 nm, c = 0.34 g/100 ml, Chloroform
LC-MS (Methode 2): Rt = 1.02 min; MS (ESIpos): m/z = 552/554 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.687 (13.49), 2.165 (16.00), 2.764 (2.32), 2.804 (2.63), 3.286 (2.32), 3.327 (2.05), 4.002 (0.44), 4.017 (0.51 ), 4.035 (0.80), 4.050 (0.75), 4.1 12 (0.80), 4.126 (0.85), 4.145 (0.53), 4.160 (0.46), 7.287 (1.94), 7.299 (2.01 ), 7.307 (2.08), 7.319 (2.1 1 ), 7.492 (1.19), 7.506 (3.59), 7.512 (2.74), 7.525 (5.63), 7.537 (7.08), 7.556 (1.33), 7.710 (3.50), 7.715 (3.73), 7.831 (2.13), 7.834 (2.36), 7.841 (1.75), 7.847 (1.73), 7.850 (2.26), 7.854 (2.20), 7.864 (2.53), 7.869 (2.38), 7.935 (4.31 ), 7.957 (2.71 ), 8.460 (2.10), 8.464 (2.24), 8.472 (2.13), 8.475 (2.11 ), 8.681 (0.95), 8.696 (1.97), 8.71 1 (0.92).
Beispiel 87
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-chlor-6-fluorphenyl)- butansäure (Racemat)
Zu einer Lösung aus (+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3- (2-chlor-6-fluorphenyl)butanoat (110 mg, 180 μηιοΙ, Beispiel 188A) in Dichlormethan (2.9 ml) wurde TFA (1.4 ml, 21 mmol) gegeben und das Gemisch wurde über Nacht bei RT gerührt. Anschlie- ßend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde in Dichlormethan aufgenommen und mittels präparativer HPLC (Methode 22) gereinigt. Es wurden 48 mg (94% Reinheit, 45% d. Th.) der Titelverbindung erhalten.
Trennung der Enantiomere:
Die Titelverbindung (45 mg) wurde in einem Gemisch aus 2 ml Isopropanol und 1 ml Heptan ge- löst und mittels präparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 88 und 89) [Säule: YMC Chiralart Amylose SA, 5 μηι, 250 mm x 30 mm; Fluss: 30 ml/min; De- tektion: 220 nm; Temperatur: 35 °C; Injektion: 1.0 ml; Eluent: 50% Heptan / 50% Isopropanol + 0.2% Essigsäure; isokratisch, Laufzeit 15 min].
LC-MS (Methode 1 ): Rt = 1.90 min; MS (ESIpos): m/z = 555/557 [M+H]+
1H-NMR (600 MHz, DMSO-d6) δ [ppm]: 0.005 (0.97), 2.143 (1.80), 2.614 (0.67), 2.642 (0.71 ), 2.735 (1.39), 2.747 (1.46), 2.760 (1.05), 2.772 (0.93), 3.715 (1.03), 4.092 (1.28), 7.179 (1.36), 7.189 (1.47), 7.203 (1.20), 7.302 (3.61 ), 7.476 (0.76), 7.479 (0.91 ), 7.481 (1.01 ), 7.484 (1.22), 7.490 (3.37), 7.492 (2.50), 7.496 (3.56), 7.501 (4.86), 7.505 (5.03), 7.506 (5.19), 7.507 (4.18), 7.510 (3.15), 7.515 (3.89), 7.519 (12.20), 7.528 (13.47), 7.530 (16.00), 7.537 (2.27), 7.541 (2.72), 7.545 (1.44), 7.839 (3.12), 7.843 (2.91 ), 7.854 (4.1 1 ), 7.858 (3.81 ), 7.927 (1.63), 7.930 (8.57), 7.945 (6.15), 9.021 (2.03).
Beispiel 88
(-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-chlor-6-fluorphenyl)butan- säure (Enantiomer 1)
Methode A:
Bei der in Beispiel 87 beschriebenen Enantiomerentrennung wurde die vorgereinigte Titelverbindung als früher eluierendes Enantiomer erhalten. Es wurden 17 mg (98% Reinheit, ee-Wert 100%) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.91 min; MS (ESIpos): m/z = 555 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (3.47), 0.008 (3.66), 2.154 (5.55), 2.709 (0.71 ), 2.749 (1.00), 2.799 (1.62), 2.814 (1.64), 2.835 (0.89), 3.314 (3.08), 3.747 (1.19), 3.873 (0.79), 4.109 (1.36), 7.175 (1.30), 7.187 (1.80), 7.199 (1.84), 7.209 (1.55), 7.227 (1.49), 7.318 (5.52),
7.476 (0.85), 7.488 (1.94), 7.491 (2.00), 7.501 (6.32), 7.509 (5.74), 7.521 (16.00), 7.526 (15.04), 7.530 (1 1.76), 7.534 (9.53), 7.537 (7.36), 7.546 (2.01 ), 7.837 (3.01 ), 7.842 (2.69), 7.860 (4.62),
7.865 (4.27), 7.931 (8.32), 7.953 (5.14), 8.940 (1.44), 8.955 (2.61 ), 8.968 (1.41 ).
Methode B:
Zu einer Lösung aus (-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3- (2-chlor-6-fluorphenyl)butanoat (1.83 g, 2.99 mmol, Beispiel 189A) in Dichlormethan (15 ml) wurde TFA (5.1 ml, 66 mmol) gegeben und das Gemisch wurde 4 h bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde in Acetonitril (9 ml) gelöst und mittels praparativer HPLC (Methode 16) gereinigt. Es wurden 1.55 g (100% Reinheit, ee-Wert 99%, 93% d. Th.) der Titelverbindung erhalten. [a]D 20 = -40.1 °, 589 nm, c = 0.48 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.02 min; MS (ESIpos): m/z = 555/557 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (4.94), 0.008 (2.48), 2.073 (0.51 ), 2.153 (5.66), 2.327 (0.54), 2.366 (0.42), 2.523 (2.16), 2.669 (0.60), 2.709 (0.68), 2.774 (1.05), 2.804 (2.40), 2.820 (2.36), 2.842 (1.06), 2.859 (0.85), 3.532 (0.98), 3.747 (1.25), 3.877 (0.89), 4.094 (1.31 ), 4.1 12 (1.61 ), 7.178 (1.12), 7.189 (1.95), 7.202 (1.90), 7.21 1 (1.52), 7.229 (1.52), 7.319 (5.49),
7.477 (1.27), 7.488 (2.48), 7.492 (2.57), 7.502 (6.85), 7.510 (6.25), 7.522 (16.00), 7.526 (14.76), 7.534 (8.66), 7.546 (1.78), 7.839 (3.24), 7.844 (2.82), 7.861 (4.86), 7.866 (4.40), 7.931 (8.73), 7.954 (5.30), 8.928 (1.89), 8.943 (3.44), 8.958 (1.69), 12.223 (0.44).
Beispiel 89
(+)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-chlor-6-fluorphenyl)butan- säure (Enantiomer 2)
Methode A:
Bei der in Beispiel 87 beschriebenen Enantiomerentrennung wurde die Titelverbindung als später eluierendes Enantiomer erhalten. Es wurden 16 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.91 min; MS (ESIpos): m/z = 555/557 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (3.94), 0.008 (3.80), 1.234 (0.72), 2.154 (5.67), 2.523 (1.40), 2.669 (0.59), 2.709 (0.81 ), 2.805 (1.47), 3.312 (3.14), 3.744 (1.07), 3.877 (0.86), 4.108 (1.32), 7.174 (0.95), 7.186 (1.80), 7.199 (1.82), 7.208 (1.52), 7.226 (1.46), 7.317 (5.76), 7.476 (0.85), 7.487 (1.93), 7.491 (2.01 ), 7.501 (6.44), 7.509 (5.74), 7.521 (16.00), 7.525 (15.01 ), 7.529 (1 1.89), 7.533 (9.55), 7.537 (7.67), 7.545 (2.01 ), 7.837 (3.09), 7.842 (2.90), 7.859 (4.69), 7.864 (4.64), 7.930 (9.07), 7.952 (5.56), 8.962 (2.38).
Methode B:
Zu einer Lösung aus (+)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3- (2-chlor-6-fluorphenyl)butanoat (1.79 g, 2.93 mmol, Beispiel 190A) in Dichlormethan (15 ml) wurde TFA (5.0 ml, 64 mmol) gegeben und das Gemisch wurde 4 h bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde in Acetonitril (9 ml) gelöst und mittels praparativer HPLC (Methode 16) gereinigt. Es wurden 1.47 g (100% Reinheit, ee-Wert 99%, 90% d. Th.) der Titelverbindung erhalten. [a]D 20 = +38.0°, 589 nm, c = 0.51 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.02 min; MS (ESIpos): m/z = 555/557 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 2.065 (0.41 ), 2.147 (5.69), 2.319 (0.45), 2.661 (0.44), 2.702 (0.53), 2.753 (0.97), 2.797 (2.33), 2.813 (2.38), 2.836 (1.06), 2.852 (0.86), 3.740 (1.43), 3.869 (0.97), 4.086 (1.28), 4.104 (1.61 ), 5.746 (0.44), 7.170 (1.01 ), 7.182 (1.87), 7.194 (1.84), 7.204 (1.52), 7.221 (1.56), 7.312 (5.74), 7.469 (0.80), 7.484 (1.96), 7.495 (6.27), 7.502 (5.58), 7.514 (16.00), 7.518 (15.07), 7.526 (9.10), 7.538 (1.91 ), 7.831 (3.13), 7.836 (2.84), 7.854 (4.76), 7.859 (4.60), 7.924 (8.90), 7.947 (5.47), 8.921 (1.79), 8.936 (3.47), 8.951 (1.73).
Beispiel 90
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(4-methylphenyl)butansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 3-(4-methylphenyl)butanoat (1 10 mg, 192 μηιοΙ, Beispiel 191A) in Dichlormethan (3.1 ml) wurde TFA (1.5 ml, 19 mmol) gegeben und das Gemisch wurde 2 h bei RT gerührt. Anschließend wur- de das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 22) gereinigt. Es wurden 98 mg (98% Reinheit, 97% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.95 min; MS (ESIpos): m/z = 517/519 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.65), 0.008 (1.37), 2.072 (1.21 ), 2.130 (2.31 ), 2.259 (16.00), 2.569 (2.41 ), 2.694 (2.27), 2.708 (2.62), 2.733 (1.79), 2.747 (1.64), 3.354 (1.01 ), 3.369 (1.50), 3.384 (1.08), 3.588 (1.02), 3.731 (0.99), 3.947 (1.29), 7.1 15 (5.01 ), 7.134 (7.42), 7.210 (9.62), 7.230 (6.30), 7.477 (0.60), 7.489 (1.53), 7.492 (1.47), 7.498 (1.78), 7.502 (5.05), 7.51 1 (4.75), 7.520 (10.92), 7.522 (12.64), 7.527 (1 1.67), 7.531 (9.46), 7.535 (7.34), 7.539 (5.85), 7.547 (1.72), 7.834 (2.13), 7.839 (2.04), 7.856 (3.30), 7.861 (3.25), 7.929 (7.08), 7.951 (4.47), 8.786 (1.42), 8.800 (2.36), 8.815 (1.44).
Trennung der Enantiomere:
Die Titelverbindung (70 mg) wurde in Acetonitril (2 ml) und Ethanol (1 ml) gelöst und mittels präparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 91 und 92) [Säule: Daicel Chiralpak IF, 5 μηη, 250 mm x 20 mm; Fluss: 20 ml/min; Detektion: 220 nm; Tempera- tur: 23 °C; Injektion: 0.08 ml; Eluent: 70% Heptan / 30% Ethanol + 0.2% TFA; isokratisch].
Beispiel 91
(+)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(4-methylphenyl)butansäure (Enantiomer 1)
Bei der in Beispiel 90 beschriebenen Enantiomerentrennung wurde die Titelverbindung als früher eluierendes Enantiomer erhalten. Es wurden 35 mg (98% Reinheit, ee-Wert 100%) der Titelverbindung erhalten.
[a]D 20 = +44.9°, 589 nm, c = 0.25 g/100 ml, Chloroform
LC-MS (Methode 1 ): Rt = 1.93 min; MS (ESIpos): m/z = 517/519 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 2.134 (2.36), 2.259 (16.00), 2.569 (2.40), 2.694 (2.27), 2.708 (2.65), 2.734 (1.74), 2.748 (1.62), 2.814 (0.62), 3.354 (0.99), 3.369 (1.51 ), 3.384 (1.07), 3.589 (0.93), 3.684 (0.89), 3.723 (0.74), 7.1 15 (4.97), 7.135 (7.41 ), 7.21 1 (9.48), 7.231 (6.28), 7.490 (1.38), 7.494 (1.39), 7.499 (1.69), 7.503 (4.96), 7.511 (4.48), 7.523 (12.30), 7.528 (1 1.39), 7.532 (9.50), 7.536 (7.18), 7.540 (5.79), 7.547 (1.75), 7.835 (2.01 ), 7.840 (1.94), 7.857 (3.21 ), 7.863 (3.16), 7.930 (6.63), 7.952 (4.20), 8.787 (1.43), 8.802 (2.41 ), 8.816 (1.48).
Beispiel 92
(-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(4-methylphenyl)butansäure (Enantiomer 2)
Bei der in Beispiel 90 beschriebenen Enantiomerentrennung wurde die Titelverbindung als später eluierendes Enantiomer erhalten. Es wurden 32 mg (98% Reinheit, ee-Wert 97%) der Titelverbindung erhalten.
[a]D 20 = -39.1 °, 589 nm, c = 0.27 g/100 ml, Chloroform
LC-MS (Methode 1 ): Rt = 1.93 min; MS (ESIpos): m/z = 517/519 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.83), 2.132 (2.34), 2.259 (16.00), 2.569 (2.38), 2.694 (2.21 ), 2.708 (2.59), 2.733 (1.71 ), 2.747 (1.62), 3.354 (0.97), 3.368 (1.46), 3.383 (1.04), 3.588 (0.93), 3.683 (0.61 ), 3.723 (0.76), 7.1 14 (5.04), 7.134 (7.42), 7.210 (9.44), 7.230 (6.18), 7.489 (1.44), 7.493 (1.47), 7.503 (5.14), 7.51 1 (4.63), 7.523 (12.42), 7.527 (1 1.59), 7.531 (9.52), 7.535 (7.23), 7.539 (5.67), 7.547 (1.67), 7.835 (2.1 1 ), 7.840 (1.98), 7.857 (3.28), 7.862 (3.18), 7.929 (6.98), 7.952 (4.34), 8.787 (1.42), 8.801 (2.34), 8.815 (1.44).
Beispiel 93
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(3-methylphenyl)butansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-
3- (3-methylphenyl)butanoat (100 mg, 174 μηιοΙ, Beispiel 192A) in Dichlormethan (2.8 ml) wurde TFA (1.3 ml, 17 mmol) gegeben und das Gemisch wurde über Nacht bei RT gerührt. Anschlie- ßend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 22) gereinigt. Es wurden 90 mg (98% Reinheit, 98% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.94 min; MS (ESIpos): m/z = 517/519 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.53), 0.008 (1.08), 2.073 (0.88), 2.1 10 (1.56), 2.267 (16.00), 2.572 (1.71 ), 2.595 (1.70), 2.705 (1.63), 2.719 (1.79), 2.744 (1.20), 2.759 (1.12), 3.356 (0.74), 3.370 (1.19), 3.385 (0.80), 3.393 (0.68), 3.594 (0.83), 3.725 (0.73), 7.025 (1.29), 7.043 (1.52), 7.1 15 (1.56), 7.138 (4.98), 7.184 (2.01 ), 7.202 (2.80), 7.221 (1.07), 7.490 (0.94), 7.493 (1.09), 7.503 (3.35), 7.51 1 (3.25), 7.522 (10.48), 7.525 (9.73), 7.530 (5.87), 7.534 (4.51 ), 7.537 (3.92), 7.545 (0.98), 7.839 (1.47), 7.844 (1.38), 7.861 (2.28), 7.867 (2.20), 7.932 (4.64), 7.954 (2.89), 8.789 (1.02), 8.804 (1.57), 8.817 (1.00).
Trennung der Enantiomere:
Die Titelverbindung (70 mg) wurde in Acetonitril (1 ml) und Ethanol (1 ml) gelöst und mittels praparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 94 und 95) [Säule: Daicel Chiralpak IF, 5 μηη, 250 mm x 20 mm; Fluss: 20 ml/min; Detektion: 220 nm; Tempera- tur: 23 °C; Injektion: 0.05 ml; Eluent: 70% Heptan / 30% Ethanol + 0.2% TFA; isokratisch].
Beispiel 94
4- {[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(3-methylphenyl)butansäure (Enantiomer 1)
Bei der in Beispiel 93 beschriebenen Enantiomerentrennung wurde die Titelverbindung als früher eluierendes Enantiomer erhalten. Es wurden 13 mg (98% Reinheit, ee-Wert 100%) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.93 min; MS (ESIpos): m/z = 517/519 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 2.109 (1.50), 2.267 (16.00), 2.572 (1.68), 2.595 (1.66), 2.705 (1.59), 2.719 (1.76), 2.745 (1.18), 2.759 (1.1 1 ), 3.355 (0.72), 3.370 (1.16), 3.385 (0.79), 3.594 (0.84), 3.724 (0.77), 7.025 (1.25), 7.043 (1.50), 7.1 15 (1.50), 7.138 (4.89), 7.184 (2.00), 7.202 (2.79), 7.221 (1.06), 7.489 (0.88), 7.492 (1.04), 7.502 (3.30), 7.510 (3.15), 7.521 (10.42), 7.524 (9.62), 7.529 (5.84), 7.533 (4.56), 7.536 (3.97), 7.544 (1.00), 7.837 (1.50), 7.842 (1.40), 7.860 (2.31 ), 7.865 (2.25), 7.931 (4.75), 7.953 (2.97), 8.787 (0.98), 8.803 (1.52), 8.816 (1.00).
Beispiel 95
4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(3-methylphenyl)butan
(Enantiomer 2)
Bei der in Beispiel 93 beschriebenen Enantiomerentrennung wurde die Titelverbindung als später eluierendes Enantiomer erhalten. Es wurden 21 mg (98% Reinheit, ee-Wert 98%) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.93 min; MS (ESIpos): m/z = 517/519 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 2.109 (1.56), 2.268 (16.00), 2.573 (1.67), 2.596 (1.67), 2.705 (1.58), 2.720 (1.76), 2.745 (1.17), 2.759 (1.1 1 ), 3.356 (0.73), 3.371 (1.19), 3.385 (0.81 ), 3.393 (0.69), 3.581 (0.76), 3.595 (0.86), 3.724 (0.78), 3.741 (0.79), 7.025 (1.28), 7.043 (1.55), 7.1 15 (1.52), 7.138 (5.00), 7.184 (1.98), 7.202 (2.76), 7.221 (1.06), 7.489 (0.87), 7.492 (1.06), 7.502 (3.29), 7.510 (3.10), 7.521 (10.40), 7.524 (10.04), 7.529 (6.10), 7.533 (4.63), 7.536 (4.10), 7.544 (1.01 ), 7.837 (1.46), 7.843 (1.38), 7.860 (2.24), 7.865 (2.23), 7.931 (4.71 ), 7.954 (2.93), 8.788 (1.01 ), 8.803 (1.56), 8.816 (1.01 ).
Beispiel 96
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-methoxyphenyl)butan- säure (Racemat)
Zu einer Lösung aus (+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3- (2-methoxyphenyl)butanoat (4.50 g, 7.63 mmol, Beispiel 193A) in Dichlormethan (52 ml) wurde TFA (12 ml, 150 mmol) gegeben und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder einge- engt. Der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (100 g Kieselgel Biotage Snap-Cartridge KP-Sil, Cyclohexan/Ethylacetat 8:2, Isolera One) gereinigt. Es wurden 3.71 g (91 % Reinheit, 83% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.88 min; MS (ESIpos): m/z = 533/535 [M+H]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.157 (1.09), 1.174 (2.15), 1.192 (1.09), 1.229 (0.95), 1.910 (0.47), 1.988 (4.22), 2.136 (10.90), 2.614 (0.49), 2.632 (0.53), 2.654 (4.30), 2.662 (4.68), 2.673 (5.14), 2.677 (5.09), 2.701 (0.49), 3.171 (16.00), 3.632 (0.75), 3.645 (1.57), 3.660 (1.69), 3.676 (1.97), 3.689 (1.60), 3.714 (1.94), 3.732 (2.38), 3.748 (1.77), 3.765 (0.90), 3.785 (1.99),
3.831 (1.42), 3.849 (0.62), 3.965 (1.62), 4.021 (1.00), 4.039 (1.01 ), 6.896 (2.08), 6.915 (4.52), 6.933 (2.59), 6.975 (4.42), 6.996 (5.33), 7.196 (2.20), 7.215 (3.50), 7.237 (5.1 1 ), 7.256 (3.76), 7.476 (0.67), 7.488 (1.83), 7.501 (6.30), 7.509 (5.32), 7.521 (14.60), 7.526 (14.79), 7.687 (2.41 ),
7.832 (3.07), 7.837 (2.72), 7.854 (4.66), 7.860 (4.45), 7.928 (8.53), 7.950 (5.31 ), 8.737 (1.87), 8.751 (3.56), 8.765 (1.87).
Trennung der Enantiomere:
Die Titelverbindung (705 mg, 100% Reinheit, erhalten aus einem anderen Experiment) wurde in einem Gemisch aus Methanol und Acetonitril (25 ml) gelöst und mittels präparativer SFC an chi- raler Phase in die Enantiomere getrennt (siehe Beispiele 97 und 98) [Säule: Daicel Chiralpak AD-H, 5 μηι, 250 mm x 20 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 0.50 ml; Eluent: 70% Kohlendioxid / 30% Isopropanol; isokratisch].
Beispiel 97
(-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-methoxyphenyl)butansäure (Enantiomer 1)
Bei der in Beispiel 96 beschriebenen Enantiomerentrennung wurde die Titelverbindung als früher eluierendes Enantiomer erhalten. Es wurden 269 mg (100% Reinheit, ee-Wert 100%) der Titelverbindung erhalten. [a]D 20 = -16.9°, 589 nm, c = 0.35 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.87 min; MS (ESIpos): m/z = 533/535 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.030 (0.91 ), 1.045 (0.92), 2.132 (4.61 ), 2.649 (1.95), 2.657 (2.08), 2.668 (2.36), 2.673 (2.28), 3.640 (0.71 ), 3.654 (0.75), 3.670 (0.90), 3.709 (0.87), 3.727 (1.06), 3.742 (0.78), 3.779 (0.81 ), 3.800 (16.00), 3.810 (1.41 ), 6.893 (0.92), 6.912 (1.98),
6.931 (1.13), 6.974 (1.95), 6.995 (2.36), 7.191 (0.90), 7.195 (0.95), 7.214 (1.50), 7.234 (2.59), 7.252 (1.72), 7.256 (1.33), 7.485 (0.82), 7.489 (0.86), 7.498 (2.75), 7.506 (2.38), 7.518 (6.93), 7.523 (6.62), 7.527 (4.73), 7.531 (3.78), 7.535 (2.89), 7.542 (0.83), 7.680 (0.94), 7.828 (1.30), 7.834 (1.13), 7.850 (2.02), 7.856 (1.84), 7.924 (3.70), 7.946 (2.35), 8.732 (0.81 ), 8.746 (1.51 ), 8.760 (0.78), 12.043 (1.22).
Beispiel 98
(+)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-methoxyph
(Enantiomer 2)
Bei der in Beispiel 96 beschriebenen Enantiomerentrennung wurde die Titelverbindung als spä- ter eluierendes Enantiomer erhalten. Es wurden 253 mg (100 % Reinheit, ee-Wert 100%) der Titelverbindung erhalten.
[a]D 20 = +18.1 °, 589 nm, c = 0.37 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.87 min; MS (ESIpos): m/z = 533/535 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 2.073 (1.06), 2.132 (4.23), 2.648 (1.75), 2.657 (1.88), 2.667 (2.13), 2.673 (2.12), 3.670 (0.82), 3.709 (0.79), 3.727 (0.97), 3.800 (16.00), 6.895 (0.85), 6.912 (1.73), 6.914 (1.83), 6.931 (1.02), 6.933 (1.02), 6.976 (1.80), 6.995 (2.17), 7.191 (0.81 ), 7.195 (0.95), 7.215 (1.42), 7.234 (2.38), 7.237 (1.73), 7.252 (1.59), 7.256 (1.29), 7.485 (0.72), 7.499 (2.50), 7.507 (2.26), 7.519 (6.59), 7.523 (6.36), 7.527 (4.56), 7.531 (3.65), 7.535 (3.04), 7.543 (0.82), 7.680 (0.87), 7.828 (1.33), 7.834 (1.18), 7.851 (2.04), 7.856 (1.96), 7.924 (3.80), 7.946 (2.41 ), 8.748 (1.34), 12.046 (0.84).
Beispiel 99
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-[2-(trifluormethoxy)phenyl]- butansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 3-[2-(trifluormethoxy)phenyl]butanoat (350 mg, 98% Reinheit, 533 mol, Beispiel 194A) in Dich- lormethan (3.8 ml) wurde TFA (820 μΙ, 1 1 mmol) gegeben und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 16) gereinigt. Es wurden 241 mg (98% Reinheit, 75% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.06 min; MS (ESIpos): m/z = 587/589 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 2.126 (4.63), 2.328 (0.57), 2.585 (1.54), 2.604 (1.66), 2.625 (2.64), 2.643 (2.55), 2.669 (0.52), 2.747 (2.51 ), 2.762 (2.75), 2.787 (1.77), 2.802 (1.67), 3.643 (1.24), 3.658 (2.05), 3.671 (1.64), 3.808 (4.26), 7.343 (3.31 ), 7.378 (6.65), 7.387 (5.79), 7.393 (5.65), 7.402 (4.92), 7.477 (0.91 ), 7.489 (2.29), 7.503 (7.37), 7.510 (6.29), 7.522 (16.00), 7.529 (15.33), 7.589 (3.82), 7.598 (3.69), 7.603 (3.76), 7.612 (3.40), 7.836 (3.22), 7.841 (3.02), 7.858 (4.95), 7.863 (4.80), 7.932 (9.52), 7.955 (5.98), 8.870 (3.68), 8.884 (1.99).
Trennung der Enantiomere:
Die Titelverbindung (210 mg) wurde in Ethanol (5 ml) gelöst und mittels praparativer HPLC an chi- raler Phase in die Enantiomere getrennt (siehe Beispiele 100 und 101 ) [Säule: Daicel Chiralcel OX-H, 5 μηη, 250 mm x 20 mm; Fluss: 15 ml/min; Detektion: 220 nm; Temperatur: 30 °C; Injektion: 0.15 ml; Eluent: 85% Heptan / 15% Ethanol; Laufzeit 1 1 min, isokratisch]. Das später eluierende Enantiomer wurde ein weiteres Mal unter den gleichen Bedingungen nachgereinigt. Die vereinig- ten Zielfraktionen wurden jeweils eingeengt und der Rückstand anschließend lyophilisiert.
Beispiel 100
(-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-[2-(trifluormethoxy)phenyl]- butansäure (Enantiomer 1)
Bei der in Beispiel 99 beschriebenen Enantiomerentrennung wurde die Titelverbindung als früher eluierendes Enantiomer erhalten. Es wurden 87 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung erhalten.
[a]D 20 = -20.0°, 589 nm, c = 0.25 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.08 min; MS (ESIpos): m/z = 587/589 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.23), 0.008 (1.04), 2.125 (4.52), 2.327 (0.45), 2.523 (1.23), 2.585 (1.75), 2.603 (1.88), 2.625 (2.85), 2.643 (2.74), 2.669 (0.49), 2.689 (3.20), 2.731 (3.78), 2.746 (2.66), 2.762 (2.95), 2.786 (1.95), 2.802 (1.83), 2.890 (4.64), 3.642 (1.26), 3.657 (2.09), 3.672 (1.71 ), 3.695 (0.78), 3.807 (4.36), 7.323 (1.73), 7.332 (2.60), 7.338 (2.78), 7.343 (3.32), 7.347 (2.98), 7.378 (6.83), 7.387 (5.84), 7.393 (5.69), 7.402 (5.04), 7.478 (0.93), 7.489 (2.33), 7.503 (7.47), 7.51 1 (6.32), 7.522 (16.00), 7.529 (15.26), 7.548 (2.47), 7.589 (3.95),
7.598 (3.72), 7.603 (3.77), 7.612 (3.43), 7.836 (3.30), 7.842 (3.04), 7.859 (5.1 1 ), 7.864 (4.91 ), 7.933 (9.89), 7.955 (6.62), 8.857 (1.92), 8.870 (3.75), 8.884 (1.99).
Beispiel 101
(+)-4-{[(6-Brom-3-methyl-2-phenylcN^
butansäure (Enantiomer 2)
Bei der in Beispiel 99 beschriebenen Enantiomerentrennung wurden 82 mg (98% Reinheit, ee-Wert 98%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = +22.8°, 589 nm, c = 0.35 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.08 min; MS (ESIpos): m/z = 587/589 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.25), 0.008 (1.39), 1.161 (0.66), 1.177 (0.65), 1.244 (1.05), 1.259 (1.29), 1.275 (0.74), 2.127 (4.52), 2.328 (0.47), 2.564 (0.42), 2.586 (1.73), 2.604 (1.83), 2.626 (2.78), 2.644 (2.70), 2.670 (0.56), 2.690 (4.00), 2.731 (5.60), 2.748 (2.62), 2.763 (2.89), 2.788 (1.91 ), 2.803 (1.76), 2.890 (6.54), 3.643 (1.24), 3.659 (2.02), 3.673 (1.64), 7.323 (1.84), 7.339 (2.89), 7.343 (3.48), 7.347 (3.09), 7.379 (6.99), 7.387 (5.95), 7.394 (5.83), 7.402 (5.10), 7.478 (1.00), 7.490 (2.52), 7.504 (7.67), 7.511 (6.72), 7.524 (16.00), 7.530 (15.63), 7.533 (14.30), 7.537 (10.49), 7.541 (8.03), 7.549 (2.55), 7.589 (3.94), 7.599 (3.70), 7.604 (3.78), 7.613 (3.42), 7.837 (3.50), 7.843 (3.23), 7.860 (5.31 ), 7.865 (5.18), 7.934 (10.61 ), 7.956 (7.03), 8.872 (3.62), 8.886 (1.92).
Beispiel 102
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(pyridin-2-yl)butansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 3-(pyridin-2-yl)butanoat (600 mg, 1.07 mmol, Beispiel 195A) in Dichlormethan (17 ml) wurde TFA (8.2 ml, 1 10 mmol) gegeben und das Gemisch wurde 16 h bei RT gerührt. Anschließend wurde
das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 22) gereinigt. Es wurden 90 mg (428 mg, 98% Reinheit, 78% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 0.80 min; MS (ESIneg): m/z = 504/506 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.12), 0.008 (1.81 ), 2.073 (0.81 ), 2.130 (5.34), 2.812 (0.58), 2.826 (0.71 ), 2.854 (2.56), 2.870 (3.13), 2.896 (2.33), 2.916 (0.60), 2.938 (0.66), 3.733 (1.84), 3.746 (1.91 ), 3.759 (0.84), 3.856 (0.67), 3.881 (1.38), 3.895 (1.37), 3.909 (0.97), 7.481 (0.70), 7.493 (1.88), 7.497 (2.00), 7.502 (2.63), 7.506 (6.48), 7.515 (5.92), 7.523 (15.59), 7.525 (16.00), 7.529 (15.81 ), 7.533 (1 1.80), 7.537 (7.95), 7.541 (6.53), 7.549 (2.08), 7.554 (1.06), 7.598 (1.98), 7.750 (1.84), 7.845 (3.15), 7.851 (2.83), 7.868 (4.80), 7.873 (4.55), 7.940 (9.37), 7.962 (5.85), 8.140 (1.50), 8.727 (2.88), 8.740 (2.68), 8.896 (1.70), 8.909 (2.72).
Trennung der Enantiomere:
Die Titelverbindung (400 mg) wurde in Methanol (33 ml) gelöst und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 103 und 104) [Säule: Daicel Chiral- pak AD, 5 μηη, 250 mm x 20 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 0.50 ml; Eluent: 78% Kohlendioxid / 22% Isopropanol; isokratisch].
Beispiel 103
(-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(pyridin-2-yl)butansäure (Enan- tiomer 1 )
Bei der in Beispiel 102 beschriebenen Enantiomerentrennung wurde die Titelverbindung als früher eluierendes Enantiomer erhalten. Es wurden 1 16 mg (98% Reinheit, ee-Wert 100%) der Titelverbindung erhalten.
[a]D 20 = -26.1 °, 589 nm, c = 0.38 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 0.80 min; MS (ESIneg): m/z = 504/506 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.17), 0.008 (1.18), 2.073 (0.99), 2.129 (4.97), 2.524 (1.1 1 ), 2.809 (0.67), 2.822 (0.84), 2.851 (2.87), 2.864 (3.16), 2.871 (3.04), 2.893 (2.57), 2.913 (0.72), 2.935 (0.80), 3.185 (2.38), 3.698 (2.1 1 ), 3.709 (2.99), 3.730 (1.89), 3.743 (2.02), 3.756 (0.91 ), 3.878 (1.35), 3.892 (1.30), 3.907 (1.00), 3.928 (0.48), 7.483 (0.61 ), 7.493 (1.72), 7.496 (1.89), 7.507 (6.03), 7.514 (5.84), 7.525 (16.00), 7.529 (15.78), 7.532 (1 1.69), 7.536 (7.83), 7.541 (6.61 ), 7.548 (2.10), 7.590 (1.92), 7.737 (1.76), 7.757 (1.84), 7.846 (3.09), 7.851
(2.89) , 7.868 (4.74), 7.873 (4.67), 7.939 (9.18), 7.962 (5.68), 8.131 (1.56), 8.723 (2.98), 8.736
(2.90) , 8.892 (1.71 ), 8.905 (2.63).
Beispiel 104
(+)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3^ (Enan- tiomer 2)
Bei der in Beispiel 102 beschriebenen Enantiomerentrennung wurde die Titelverbindung als spä- ter eluierendes Enantiomer erhalten. Es wurden 122 mg (98% Reinheit, ee-Wert 95%) der Titelverbindung erhalten.
[a]D 20 = +23.1 °, 589 nm, c = 0.33 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 0.80 min; MS (ESIneg): m/z = 504/506 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.06), 0.008 (1.29), 2.073 (0.85), 2.130 (4.93), 2.329 (0.40), 2.812 (0.71 ), 2.826 (0.83), 2.855 (2.89), 2.868 (3.23), 2.874 (3.13), 2.896 (2.56), 2.916 (0.72), 2.938 (0.77), 3.713 (3.01 ), 3.733 (1.91 ), 3.746 (2.07), 3.759 (0.91 ), 3.880 (1.32), 3.895 (1.32), 3.909 (0.97), 7.483 (0.78), 7.497 (2.06), 7.507 (6.21 ), 7.515 (6.03), 7.525 (16.00), 7.529 (15.65), 7.537 (7.59), 7.541 (6.26), 7.599 (1.99), 7.748 (1.90), 7.768 (1.91 ), 7.846 (3.1 1 ), 7.851 (2.82), 7.868 (4.67), 7.874 (4.50), 7.940 (9.06), 7.962 (5.53), 8.143 (1.66), 8.729 (3.09), 8.740 (2.92), 8.895 (1.76), 8.909 (2.62).
Beispiel 105
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-[2-(trifluormethyl)phenyl]- butansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 3-[2-(trifluormethyl)phenyl]butanoat (1.00 g, 1.59 mmol, Beispiel 196A) in Dichlormethan (15 ml) wurde TFA (2.5 ml, 32 mmol) gegeben, und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 20) ge- reinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 845 mg (98% Reinheit, 91 % d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): R
t = 1.99 min; MS (ESIpos): m/z = 571/573 [M+H]
+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.60), 0.008 (2.44), 2.1 12 (4.45), 2.328 (0.57), 2.606 (1.73), 2.622 (1.84), 2.646 (3.08), 2.662 (3.07), 2.737 (2.87), 2.755 (3.03), 2.777 (1.83), 2.795 (1.75), 3.715 (1.38), 3.787 (1.81 ), 3.804 (2.48), 3.819 (1.89), 3.898 (1.74), 7.448 (1.64), 7.468 (3.75), 7.487 (4.29), 7.501 (7.41 ), 7.508 (6.42), 7.520 (16.00), 7.526 (15.35), 7.668 (1.84), 7.686 (3.59), 7.712 (4.92), 7.732 (3.88), 7.776 (3.72), 7.796 (2.64), 7.826 (3.45), 7.832 (3.21 ), 7.849 (5.09), 7.854 (5.02), 7.926 (9.89), 7.948 (6.33), 8.856 (1.93), 8.871 (3.79), 8.885 (1.87).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 12.20 (br. s, 1 H), 8.87 (t, 2H), 7.93 (d, 1 H), 7.84 (dd, 1 H), 7.79 (d, 1 H), 7.75-7.56 (m, 3H), 7.56-7.44 (m, 6H), 3.93-3.85 (br. m, 1 H), 3.84-3.76 (m, 1 H), 3.75-3.65 (br. m, 1 H), 2.76 (dd, 1 H), 2.64 (dd, 1 H), 2.1 1 (br. s, 3H).
Trennung der Enantiomere:
Die Titelverbindung (750 mg) wurde in einem 1 :1 -Gemisch aus Acetonitril und Methanol (60 ml) gelöst und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 106 und 107) [Säule: Chiralpak AD-H, 5 μηι, 250 mm x 30 mm; Fluss: 100 ml/min; Detek- tion: 210 nm; Temperatur: 38 °C; Injektion: 2.2 ml; Eluent: 75% Kohlendioxid / 25% Ethanol; Laufzeit 15.5 min, isokratisch].
Beispiel 106
(+)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-[2-(trifluormethyl)phenyl]- butansäure (Enantiomer 1)
Bei der in Beispiel 105 beschriebenen Enantiomerentrennung wurden 363 mg (98% Reinheit, ee-Wert 100%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = +31.6°, 589 nm, c = 0.47 g/100 ml, Methanol
Anschließend wurden enthaltende Ethanol-Reste durch Lyophilisation entfernt.
LC-MS (Methode 1 ): Rt = 2.00 min; MS (ESIpos): m/z = 571/573 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.38), 0.008 (1.00), 1.234 (0.45), 2.1 13 (4.47), 2.328 (0.49), 2.607 (1.66), 2.623 (1.82), 2.646 (2.93), 2.663 (2.93), 2.710 (0.52), 2.737 (2.83), 2.755 (2.99), 2.777 (1.82), 2.795 (1.69), 3.710 (1.21 ), 3.786 (1.68), 3.803 (2.20), 3.819 (1.64), 3.883 (1.19), 7.449 (1.73), 7.469 (3.93), 7.487 (4.52), 7.500 (7.72), 7.508 (6.99), 7.520 (16.00), 7.526 (15.16), 7.533 (10.45), 7.545 (2.74), 7.668 (1.89), 7.687 (3.63), 7.713 (4.86), 7.733 (3.86), 7.776 (3.76), 7.796 (2.57), 7.825 (3.46), 7.830 (3.27), 7.847 (5.01 ), 7.853 (4.91 ), 7.925 (9.61 ), 7.948 (6.13), 8.856 (2.04), 8.871 (3.74), 8.885 (1.85), 12.218 (3.06).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 12.22 (br. s, 1 H), 8.87 (t, 1 H), 7.93 (d, 1 H), 7.84 (dd, 1 H), 7.79 (d, 1 H), 7.75-7.56 (m, 3H), 7.56-7.43 (m, 6H), 3.95-3.85 (br. m, 1 H), 3.85-3.76 (m, 1 H), 3.75-3.65 (br. m, 1 H), 2.76 (dd, 1 H), 2.64 (dd, 1 H), 2.1 1 (br. s, 3H).
Beispiel 107
(-)-4-{[(6-Brom-3-methyl-2-phenylchi^
butansäure (Enantiomer 2)
Bei der in Beispiel 105 beschriebenen Enantiomerentrennung wurden 357 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = -27.3°, 589 nm, c = 0.46 g/100 ml, Methanol
Anschließend wurden enthaltende Ethanol-Reste durch Lyophilisation entfernt.
LC-MS (Methode 1 ): Rt = 2.00 min; MS (ESIpos): m/z = 571/573 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.149 (0.67), -0.008 (7.44), 0.008 (5.88), 0.146 (0.72), 1.234 (0.59), 2.1 13 (4.41 ), 2.327 (0.78), 2.602 (1.69), 2.618 (1.67), 2.642 (2.92), 2.658 (2.90), 2.669 (1.08), 2.734 (2.79), 2.752 (2.96), 2.774 (1.85), 2.792 (1.75), 3.714 (1.21 ), 3.783 (1.64), 3.801 (2.17), 3.818 (1.64), 3.881 (1.16), 7.449 (1.66), 7.467 (3.70), 7.486 (4.16), 7.500 (7.27), 7.508 (6.39), 7.520 (16.00), 7.525 (15.08), 7.533 (10.41 ), 7.545 (2.74), 7.667 (1.78), 7.686 (3.55), 7.712 (4.86), 7.732 (3.84), 7.774 (3.71 ), 7.794 (2.60), 7.825 (3.38), 7.830 (3.12), 7.847 (5.02), 7.852 (4.86), 7.924 (9.40), 7.947 (6.02), 8.867 (1.72), 8.882 (3.06), 12.220 (0.72).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 12.22 (br. s, 1 H), 8.88 (t, 1 H), 7.93 (d, 1 H), 7.84 (dd, 1 H), 7.78 (d, 1 H), 7.76-7.58 (m, 3H), 7.56-7.43 (m, 6H), 3.95-3.85 (br. m, 1 H), 3.84-3.76 (m, 1 H), 3.75-3.64 (br. m, 1 H), 2.76 (dd, 1 H), 2.64 (dd, 1 H), 2.1 1 (br. s, 3H).
Beispiel 108
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(3-methoxypyridin-2-yl)butan- säure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 3-(3-methoxypyridin-2-yl)butanoat (1.70 g, 2.88 mmol, Beispiel 197A) in Dichlormethan (47 ml) wurde TFA (22 ml, 290 mmol) gegeben und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan ver- setzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 22) gereinigt. Es wurden 1.28 g (98% Reinheit, 82% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.61 min; MS (ESIpos): m/z = 534/536 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.96), 0.008 (0.66), 2.189 (16.00), 2.607 (1.12), 2.622 (1.19), 2.648 (1.52), 2.662 (1.44), 2.880 (1.29), 2.902 (1.37), 2.921 (1.06), 2.942 (1.03), 3.170 (1.45), 3.342 (0.58), 3.709 (1.46), 3.722 (1.52), 3.957 (0.99), 3.972 (1.05), 3.978 (1.07), 3.994 (0.83), 7.224 (1.46), 7.236 (1.54), 7.245 (1.83), 7.257 (1.81 ), 7.384 (2.13), 7.403 (1.63), 7.477 (0.46), 7.489 (1.34), 7.504 (3.55), 7.510 (2.87), 7.522 (5.70), 7.536 (6.77), 7.553 (1.24), 7.763 (2.16), 7.836 (1.69), 7.842 (1.42), 7.858 (2.54), 7.864 (2.29), 7.930 (4.36), 7.952 (2.72), 8.1 16 (2.25), 8.1 19 (2.29), 8.128 (2.22), 8.130 (2.07), 8.730 (0.98), 8.745 (1.93), 8.760 (0.89), 1 1.987 (0.59).
Trennung der Enantiomere:
Die Titelverbindung (1.20 g) wurde in 45 ml eines Gemisches aus Ethanol, Heptan und Dichlormethan warm gelöst und mittels präparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 109 und 1 10) [Säule: YMC Chiralart Amylose SA, 5 μηι, 250 mm x 30 mm; Fluss: 50 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 0.60 ml; Eluent: 60% Heptan / 40% Isopropanol; isokratisch; Laufzeit 7 min].
Beispiel 109
(-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(3-methoxypyridin-2-yl)- butansäure (Enantiomer 1)
Bei der in Beispiel 108 beschriebenen Enantiomerentrennung wurden 526 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -14.7°, 589 nm, c = 0.50 g/100 ml, Chloroform
LC-MS (Methode 1 ): Rt = 1.60 min; MS (ESIpos): m/z = 534/536 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.031 (2.09), 1.047 (2.10), 2.191 (16.00), 2.606 (1.02), 2.621 (1.09), 2.647 (1.39), 2.661 (1.35), 2.881 (1.17), 2.903 (1.24), 2.921 (0.95), 2.943 (0.95), 3.709 (1.37), 3.723 (1.44), 3.758 (0.43), 3.958 (0.90), 3.973 (1.01 ), 3.979 (1.04), 3.995 (0.80), 7.219 (1.72), 7.231 (1.77), 7.240 (2.16), 7.252 (2.17), 7.378 (2.43), 7.397 (1.90), 7.489 (1.26), 7.504 (3.55), 7.510 (2.76), 7.523 (5.77), 7.536 (7.05), 7.554 (1.26), 7.765 (2.40), 7.836 (1.75),
7.841 (1.43), 7.858 (2.63), 7.864 (2.36), 7.930 (4.63), 7.952 (2.85), 8.1 14 (2.39), 8.1 17 (2.44), 8.126 (2.44), 8.129 (2.34), 8.142 (1.51 ), 8.731 (0.91 ), 8.745 (1.87), 8.760 (0.87).
Beispiel 110
(+)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3^
butansäure (Enantiomer 2)
Bei der in Beispiel 108 beschriebenen Enantiomerentrennung wurde die vorgereinigte Titelverbindung (90% Reinheit, ee-Wert 98%) erhalten.
[a]D 20 = +12.3°, 589 nm, c = 0.49 g/100 ml, Chloroform
Anschließend wurde die vorgereinigte Titelverbindung zweimal mittels präparativer HPLC (Me- thode 20) und einmal mittels präparativer HPLC [Säule: XBridge C18, 5 μηι, 100 mm x 30 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 0.25 ml; Eluent: 85% Wasser / 5% (Acetonitril/Wasser 8:2) + 2% Ammoniak-Lösung / 5% Acetonitril -> 75% Wasser / 5% (Aceton itril/Wasser 8:2) + 2% Ammoniak-Lösung / 20% Acetonitril; Laufzeit 5 min] nachgereinigt. Es wurden 240 mg (98% Reinheit) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 0.84 min; MS (ESIpos): m/z = 534/536 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.754 (7.46), 2.189 (16.00), 2.577 (1.13), 2.592 (1.16), 2.617 (1.47), 2.632 (1.42), 2.836 (1.15), 2.856 (1.20), 2.876 (0.95), 2.897 (0.95), 3.308 (0.85), 3.71 1 (1.82), 3.947 (1.03), 3.968 (1.21 ), 3.983 (0.92), 7.215 (1.70), 7.227 (1.79), 7.236 (2.21 ), 7.248 (2.22), 7.373 (2.57), 7.393 (1.96), 7.488 (1.23), 7.503 (3.63), 7.509 (2.81 ), 7.522 (5.99), 7.535 (7.46), 7.552 (1.37), 7.761 (2.46), 7.834 (1.63), 7.839 (1.40), 7.857 (2.52), 7.862 (2.31 ), 7.928 (4.44), 7.950 (2.72), 8.1 13 (2.53), 8.123 (2.44), 8.804 (1.24).
Beispiel 111
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-chlorphenyl)-2,2-dimethyl- butansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-
3- (2-chlorphenyl)-2,2-dimethylbutanoat (330 mg, 531 mol, Beispiel 198A) in Dichlormethan (10 ml) wurde TFA (5 ml, 64.9 mmol) gegeben und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und zweimal hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode: Chromatorex C18, 10 μΜ, 125 x 30 mm, Acetonitril/Wasser-Gradient mit 0.01 % TFA) gereinigt. Es wurden 248 mg (99% Reinheit, 82% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 7): Rt = 1.45 min; MS (ESIpos): m/z = 565/567 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.38), 0.008 (2.15), 1.099 (16.00), 1.1 15 (15.35), 1.919 (0.56), 2.073 (0.42), 2.327 (0.79), 2.366 (0.42), 2.523 (2.16), 2.670 (0.71 ), 3.661 (1.15), 3.936 (1.92), 4.098 (4.62), 7.270 (1.1 1 ), 7.365 (1.10), 7.433 (3.27), 7.453 (3.05), 7.486 (13.79), 7.491 (13.00), 7.504 (3.35), 7.524 (2.60), 7.545 (1.69), 7.798 (1.45), 7.821 (2.24), 7.888 (4.91 ), 7.91 1 (2.96), 8.696 (1.97).
1 H-NMR (400 MHz, DMSO-d6): δ [ppm] = 12.55 (br. s, 1 H), 8.75-8.62 (m, 1 H), 7.90 (d, 1 H), 7.81 (dd, 1 H), 7.72-7.18 (m, 10H), 4.14-4.03 (m, 2H, teilweise verdeckt), 3.71 -3.62 (m, 1 H, teilweise verdeckt), 1.92 (br. s, 3H), 1.12 (s, 3H), 1.10 (s, 3H).
Trennung der Enantiomere:
Die Titelverbindung (212 mg) wurde in 25 ml eines Gemisches aus Methanol und Acetonitril gelöst und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 1 12 und 1 13) [Säule: Daicel Chiralpak AD SFC, 5 μηι, 250 mm x 20 mm; Fluss: 80 ml/min; De- tektion: 210 nm; Temperatur: 40 °C; Injektion: 1.50 ml; Eluent: 70% Kohlendioxid / 30% Ethanol; isokratisch; Laufzeit 7 min].
Beispiel 112
4- {[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-chlorphenyl)-2,2-dimethyl- butansäure (Enantiomer 1)
Bei der in Beispiel 1 1 1 beschriebenen Enantiomerentrennung wurden 80 mg (99% Reinheit, ee-Wert 100%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
LC-MS (Methode 2): Rt = 1.12 min; MS (ESIpos): m/z = 565/567 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.87), 0.008 (3.23), 1.099 (16.00), 1.1 15 (15.49), 1.937 (0.56), 2.327 (0.54), 2.366 (0.44), 2.523 (1.74), 2.670 (0.55), 2.710 (0.43), 3.663 (0.94), 4.098 (4.16), 7.272 (1.12), 7.366 (1.14), 7.433 (3.29), 7.453 (3.08), 7.485 (14.05), 7.491 (13.77), 7.503 (4.00), 7.515 (1.29), 7.527 (2.52), 7.545 (1.78), 7.797 (1.48), 7.819 (2.29), 7.887 (4.96), 7.909 (3.06), 8.695 (2.05), 12.562 (3.44).
1 H-NMR (400 MHz, DMSO-d6): δ [ppm] = 12.56 (s, 1 H), 8.73-8.64 (m, 1 H), 7.90 (d, 1 H), 7.81 (dd, 1 H), 7.57-7.22 (m, 10H), 4.14-4.06 (m, 2H), 3.71-3.62 (m, 1 H), 1.94 (br. s, 3H), 1.1 1 (s, 3H), 1.10 (s, 3H).
Beispiel 113
4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-chlorph
dimethylbutansäure (Enantiomer 2)
Bei der in Beispiel 1 1 1 beschriebenen Enantiomerentrennung wurden 81 mg (99% Reinheit, ee-Wert 99%) der Titelverbindung als später eluierendes Enantiomer erhalten.
LC-MS (Methode 2): Rt = 1.12 min; MS (ESIpos): m/z = 565/567 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.88), 0.008 (2.30), 1.099 (16.00), 1.1 14 (15.33), 1.939 (0.59), 2.073 (0.45), 2.327 (0.48), 2.523 (1.54), 2.670 (0.41 ), 3.662 (0.94), 4.098 (4.20), 7.271 (1.12), 7.364 (1.1 1 ), 7.433 (3.21 ), 7.455 (2.99), 7.486 (14.01 ), 7.491 (13.51 ), 7.503 (4.03), 7.515 (1.35), 7.527 (2.61 ), 7.546 (1.84), 7.797 (1.44), 7.819 (2.27), 7.823 (2.23), 7.887 (4.73), 7.909 (2.96), 8.698 (1.95), 12.563 (1.12).
1 H-NMR (400 MHz, DMSO-d6): δ [ppm] = 12.56 (br. s, 1 H), 8.76-8.64 (m, 1 H), 7.90 (d, 1 H), 7.81 (dd, 1 H), 7.57-7.21 (m, 10H), 4.14-4.04 (m, 2H), 3.72-3.61 (m, 1 H), 1.94 (br. s, 3H), 1.1 1 (s, 3H), 1.10 (s, 3H).
Beispiel 114
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-chlorphenyl)-3-methyl- butansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 3-(2-chlorphenyl)-3-methylbutanoat (1.70 g, 87% Reinheit, 2.43 mmol, Beispiel 199A) in Di- chlormethan (17 ml) wurde TFA (3.7 ml, 49 mmol) gegeben, und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit
Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 15) gereinigt. Es wurden 1.06 g (98% Reinheit, 77% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.06 min; MS (ESIpos): m/z = 517/519 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.48), 0.008 (1.95), 1.546 (0.42), 1.672 (16.00), 2.072 (1.61 ), 2.1 12 (1.69), 2.327 (0.51 ), 2.523 (1.63), 2.669 (0.51 ), 2.760 (4.48), 2.798 (5.03), 3.342 (3.72), 3.381 (3.37), 3.777 (1.67), 3.790 (1.85), 3.81 1 (2.05), 3.824 (1.90), 4.342 (1.91 ), 4.359 (2.08), 4.375 (1.90), 4.392 (1.78), 7.229 (0.83), 7.248 (2.29), 7.267 (2.18), 7.274 (2.24), 7.278 (2.34), 7.293 (2.82), 7.297 (2.96), 7.312 (1.36), 7.392 (2.60), 7.41 1 (2.1 1 ), 7.459 (3.52), 7.463 (3.53), 7.478 (3.27), 7.482 (3.14), 7.500 (5.81 ), 7.508 (5.01 ), 7.519 (12.10), 7.526 (1 1.50), 7.544 (2.31 ), 7.654 (0.63), 7.831 (2.64), 7.836 (2.49), 7.853 (4.1 1 ), 7.859 (4.10), 7.926 (7.99), 7.949 (5.13), 8.624 (1.72), 8.640 (2.91 ), 8.655 (1.76).
Beispiel 115
(+)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-fluorphenyl)-3-methylbutan- säure (Enantiomer 1 )
Zu einer Lösung aus (+)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3- (2-fluorphenyl)-3-methylbutanoat (80 mg, 135 μηηοΙ, Beispiel 201A) in Dichlormethan (2 ml) wurde TFA (210 μΙ, 2.7 mmol) gegeben und das Gemisch wurde über Nacht bei RT gerührt. Anschlie- ßend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 16) gereinigt. Es wurden 69 mg (98% Reinheit, ee-Wert 100%, 93% d. Th.) der Titelverbindung erhalten.
[a]D 20 = +31.5°, 589 nm, c = 0.31 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.92 min; MS (ESIpos): m/z = 535/537 [M+H]+
1H-NMR (600 MHz, DMSO-d6) δ [ppm]: 1.590 (16.00), 2.076 (1.84), 2.158 (0.55), 2.652 (3.56), 2.677 (4.15), 2.968 (3.73), 2.993 (3.31 ), 3.668 (1.44), 3.677 (1.57), 3.690 (1.69), 3.699 (1.54),
4.067 (1.93), 4.079 (2.09), 4.089 (1.90), 4.101 (1.75), 7.1 18 (1.02), 7.140 (2.13), 7.153 (2.82), 7.292 (1.26), 7.350 (1.58), 7.364 (2.78), 7.376 (1.38), 7.486 (0.77), 7.494 (2.62), 7.500 (2.28),
7.505 (4.1 1 ), 7.508 (5.56), 7.521 (8.62), 7.533 (10.91 ), 7.545 (2.64), 7.843 (2.16), 7.847 (2.13), 7.858 (2.94), 7.861 (2.90), 7.936 (5.68), 7.951 (4.15), 8.715 (1.79), 8.726 (3.02), 8.736 (1.77). 1H-NMR (600 MHz, DMSO-d6): δ [ppm] = 8.73 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.66 (br. s, 1 H), 7.59-7.46 (m, 5H), 7.36 (br. t, 1 H), 7.29 (br. s, 1 H), 7.19-7.09 (m, 2H), 4.08 (dd, 1 H), 3.68 (dd, 1 H), 2.98 (d, 1 H), 2.66 (d, 1 H), 2.08 (br. s, 3H), 1.59 (s, 3H).
Beispiel 116
(-)-4-{[(6-Brom-3-methyl-2-phenylchin^
säure (Enantiomer 2)
Zu einer Lösung aus (-)-ie f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3- (2-fluorphenyl)-3-methylbutanoat (80 mg, 135 μηιοΙ, Beispiel 202A) in Dichlormethan (2 ml) wurde TFA (210 μΙ, 2.7 mmol) gegeben und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan ver- setzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 16) gereinigt. Es wurden 60 mg (98% Reinheit, ee-Wert 100%, 81 % d. Th.) der Titelverbindung erhalten.
[a]D 20 = -28.9°, 589 nm, c = 0.32 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.92 min; MS (ESIpos): m/z = 535/537 [M+H]+
1H-NMR (600 MHz, DMSO-d6) δ [ppm]: 1.587 (16.00), 2.076 (0.93), 2.649 (3.54), 2.674 (4.12), 2.965 (3.72), 2.991 (3.30), 3.664 (2.15), 3.673 (2.30), 3.686 (2.47), 3.696 (2.30), 4.063 (1.95), 4.075 (2.10), 4.086 (1.90), 4.097 (1.75), 7.1 18 (1.05), 7.139 (2.16), 7.152 (2.84), 7.291 (1.29), 7.348 (1.60), 7.361 (2.79), 7.375 (1.39), 7.484 (0.86), 7.491 (2.60), 7.498 (2.40), 7.503 (4.00),
7.506 (5.30), 7.519 (8.77), 7.530 (1 1.64), 7.542 (2.44), 7.840 (2.20), 7.855 (2.98), 7.932 (5.50), 7.947 (4.00), 8.710 (1.80), 8.720 (3.02), 8.730 (1.73).
1H-NMR (600 MHz, DMSO-d6): δ [ppm] = 12.00 (br. s, 1 H), 8.72 (t, 1 H), 7.94 (d, 1 H), 7.85 (d, 1 H), 7.64 (br. s, 1 H), 7.56-7.47 (m, 5H), 7.36 (t, 1 H), 7.28 (br. s, 1 H), 7.19-7.09 (m, 2H), 4.08 (dd, 1 H), 3.68 (dd, 1 H), 2.98 (d, 1 H), 2.66 (d, 1 H), 2.10 (br. s, 3H), 1.59 (s, 3H).
Beispiel 117
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-methyl-3-(pyridin-2-yl)butan- säure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 3-methyl-3-(pyridin-2-yl)butanoat (900 mg, 1.57 mmol, Beispiel 203A) in Dichlormethan (20 ml) wurde TFA (2.4 ml, 31 mmol) gegeben, und das Gemisch wurde über Nacht bei RT gerührt. An- schließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 16) gereinigt. Es wurden 585 mg (98% Reinheit, 71 % d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.67 min; MS (ESIpos): m/z = 518/520 [M+H]+
1H-NMR (600 MHz, DMSO-d6) δ [ppm]: 1.199 (0.59), 1.210 (0.60), 1.578 (16.00), 1.686 (0.78), 2.077 (2.22), 2.148 (2.50), 2.172 (1.84), 2.503 (14.98), 2.735 (2.77), 2.762 (3.14), 3.099 (2.31 ), 3.125 (2.03), 3.765 (1.15), 3.774 (1.28), 3.787 (1.47), 3.797 (1.32), 3.973 (1.15), 3.984 (1.24), 3.995 (1.04), 4.006 (0.93), 7.439 (1.01 ), 7.487 (0.64), 7.498 (2.33), 7.509 (3.62), 7.512 (4.88), 7.525 (6.71 ), 7.539 (3.97), 7.543 (7.16), 7.555 (3.07), 7.689 (1.32), 7.701 (1.35), 7.850 (2.35), 7.854 (2.27), 7.865 (3.07), 7.869 (2.96), 7.945 (5.52), 7.960 (4.20), 7.988 (0.95), 7.994 (0.98), 8.007 (0.53), 8.642 (1.83), 8.649 (1.81 ), 8.753 (1.14), 8.763 (2.10), 8.773 (1.14).
Trennung der Enantiomere:
Die Titelverbindung (530 mg) wurde in Methanol (30 ml) gelöst und mittels praparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 1 18 und 1 19) [Säule: Chiralpak AD- H, 5 μηη, 250 mm x 20 mm; Fluss: 50 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 0.40 ml; Eluent: 78% Kohlendioxid / 22% Isopropanol; isokratisch; Laufzeit 17 min].
Beispiel 118
(+)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-methyl-3-(pyridin-2-yl)butan- säure (Enantiomer 1)
Bei der in Beispiel 1 17 beschriebenen Enantiomerentrennung wurden 202 mg (98% Reinheit, ee-Wert 100%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = +17.8°, 589 nm, c = 0.34 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.68 min; MS (ESIpos): m/z = 518/520 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.63), 0.008 (1.38), 1.030 (2.31 ), 1.045 (2.33), 1.544 (16.00), 2.170 (15.68), 2.518 (1.12), 2.523 (0.86), 2.672 (3.09), 2.71 1 (3.64), 3.000 (3.08), 3.039 (2.56), 3.473 (1.48), 3.782 (0.75), 3.797 (0.79), 3.816 (1.38), 3.830 (1.29), 3.873 (1.33), 3.888 (1.41 ), 3.906 (0.76), 3.921 (0.68), 7.208 (1.60), 7.210 (1.67), 7.220 (1.73), 7.222 (1.90), 7.226 (1.95), 7.228 (1.80), 7.238 (1.78), 7.241 (1.76), 7.477 (0.66), 7.486 (3.63), 7.488 (3.36), 7.499 (3.31 ), 7.504 (7.89), 7.517 (2.42), 7.522 (6.46), 7.539 (8.25), 7.543 (6.47), 7.548 (3.28), 7.552 (1.58), 7.558 (1.91 ), 7.564 (1.21 ), 7.701 (2.51 ), 7.732 (1.65), 7.736 (1.73), 7.751 (2.38), 7.756 (2.43), 7.771 (1.29), 7.775 (1.27), 7.835 (2.24), 7.841 (1.98), 7.858 (3.38), 7.863 (3.19), 7.934 (5.82), 7.956 (3.76), 8.527 (2.15), 8.529 (2.21 ), 8.539 (2.09), 8.541 (1.95), 8.628 (1.1 1 ), 8.644 (2.31 ), 8.659 (1.18), 1 1.953 (4.48).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 1 1.95 (s, 1 H), 8.64 (t, 1 H), 8.53 (dd, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.75 (td, 1 H), 7.70 (s, 1 H), 7.57-7.46 (m, 6H), 7.22 (ddd, 1 H), 3.89 (dd, 1 H), 3.81 (dd, 1 H), 3.02 (d, 1 H), 2.69 (d, 1 H), 2.17 (s, 3H), 1.54 (s, 3H). Beispiel 119
(-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-methyl-3-(pyridi
säure (Enantiomer 2)
Bei der in Beispiel 1 17 beschriebenen Enantiomerentrennung wurden 72 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als später eluierendes Enantiomer erhalten. [a]D 20 = -22.1 °, 589 nm, c = 0.35 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.68 min; MS (ESIpos): m/z = 518/520 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.49), 0.008 (1.29), 1.542 (16.00), 2.171 (15.95), 2.523 (0.88), 2.673 (3.04), 2.712 (3.53), 2.995 (2.98), 3.034 (2.44), 3.786 (0.63), 3.801 (0.73), 3.819 (1.36), 3.834 (1.26), 3.870 (1.31 ), 3.885 (1.39), 3.903 (0.71 ), 3.918 (0.62), 7.209 (1.73), 7.221 (1.94), 7.226 (1.93), 7.238 (1.79), 7.485 (3.48), 7.504 (8.08), 7.522 (6.48), 7.539 (8.24), 7.542 (6.57), 7.548 (3.23), 7.558 (1.89), 7.702 (2.74), 7.731 (1.63), 7.735 (1.67), 7.750 (2.44), 7.755 (2.43), 7.770 (1.25), 7.774 (1.23), 7.835 (2.26), 7.840 (2.01 ), 7.857 (3.39), 7.863 (3.19), 7.934 (6.13), 7.956 (3.87), 8.530 (2.34), 8.539 (2.30), 8.672 (1.37), 1 1.958 (0.90).
Beispiel 120
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-methoxyphenyl)-3-methyl- butansäure (Racemat)
Zu einer Lösung aus (+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3- (2-methoxyphenyl)-3-methylbutanoat (100 mg, 98% Reinheit, 162 μηιοΙ, Beispiel 204A) in Dichlormethan (1.2 ml) wurde TFA (250 μΙ, 3.2 mmol) gegeben, und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 20) gereinigt. Es wurden 84 mg (98% Reinheit, 92% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.97 min; MS (ESIpos): m/z = 547/549 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.572 (13.20), 2.072 (2.90), 2.655 (3.43), 2.691 (3.96), 3.072 (3.56), 3.109 (3.10), 3.691 (1.52), 3.704 (1.64), 3.723 (1.81 ), 3.737 (1.66), 4.205 (1.34), 4.223 (1.47), 4.238 (1.34), 4.255 (1.22), 6.868 (1.38), 6.887 (2.95), 6.905 (1.69), 6.977 (2.51 ), 6.998 (3.00), 7.188 (1.45), 7.208 (2.41 ), 7.220 (3.81 ), 7.239 (3.02), 7.475 (0.44), 7.486 (1.36), 7.497 (4.27), 7.505 (4.46), 7.516 (16.00), 7.660 (0.69), 7.825 (2.17), 7.831 (2.05), 7.848 (3.41 ), 7.853 (3.40), 7.919 (6.21 ), 7.941 (3.90), 8.480 (1.38), 8.496 (2.48), 8.511 (1.39). Beispiel 121
(+)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-methoxyphenyl)-3-methyl- butansäure (Enantiomer 1)
Zu einer Lösung aus (+)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3- (2-methoxyphenyl)-3-methylbutanoat (650 mg, 1.08 mmol, Beispiel 205A) in Dichlormethan (16 ml) wurde TFA (1.7 ml, 22 mmol) gegeben und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 16) gereinigt. Es wurden 398 mg (98% Reinheit, ee-Wert 95%, 66% d. Th.) der Titelverbindung erhalten.
[a]D 20 = +30.0°, 589 nm, c = 0.44 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.04 min; MS (ESIpos): m/z = 547/549 [M+H]+
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: -0.007 (0.51 ), 1.254 (0.43), 1.577 (16.00), 2.071 (4.33), 2.080 (1.58), 2.664 (4.50), 2.693 (5.04), 3.080 (4.33), 3.109 (3.90), 3.702 (1.89), 3.712 (2.07), 3.728 (2.22), 3.739 (1.98), 4.215 (1.45), 4.229 (1.60), 4.241 (1.51 ), 4.255 (1.33), 6.874 (1 .58), 6.889 (3.28), 6.904 (1.84), 6.980 (2.73), 6.996 (3.14), 7.192 (1.44), 7.207 (2.39), 7.226 (4.53), 7.229 (3.80), 7.241 (3.60), 7.244 (3.13), 7.477 (0.76), 7.486 (1.89), 7.488 (1.64), 7.491 (1.52), 7.498 (5.02), 7.503 (4.86), 7.515 (1 1.97), 7.520 (1 1.66), 7.525 (10.63), 7.536 (2.01 ), 7.674 (0.59), 7.828 (3.01 ), 7.833 (2.86), 7.846 (4.20), 7.851 (4.18), 7.923 (8.09), 7.941 (5.60), 8.487 (1.74), 8.499 (2.97), 8.512 (1.75).
Beispiel 122
(-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-meth
butansäure (Enantiomer 2)
Zu einer Lösung aus (-)-ierf-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2- methoxyphenyl)-3-methylbutanoat (650 mg, 1.08 mmol, Beispiel 206A) in Dichlormethan (16 ml) wurde TFA (1.7 ml, 22 mmol) gegeben und das Gemisch wurde über Nacht bei RT gerührt. An- schließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 16) gereinigt. Es wurden 468 mg (98% Reinheit, ee-Wert 99%, 78% d. Th.) der Titelverbindung erhalten.
[a]D 20 = -35.6°, 589 nm, c = 0.43 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.04 min; MS (ESIpos): m/z = 547/549 [M+H]+
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: -0.007 (0.57), 1.575 (16.00), 2.071 (3.31 ), 2.663 (4.52), 2.692 (5.04), 3.079 (4.34), 3.108 (3.90), 3.700 (1.92), 3.71 1 (2.10), 3.726 (2.23), 3.737 (1.99), 4.213 (1.45), 4.227 (1.59), 4.239 (1.47), 4.253 (1.30), 6.873 (1.60), 6.888 (3.29), 6.903 (1 .83), 6.979 (2.75), 6.995 (3.13), 7.191 (1.48), 7.206 (2.42), 7.225 (4.71 ), 7.228 (3.78), 7.241 (3.64), 7.243 (3.02), 7.471 (0.43), 7.476 (0.86), 7.485 (1.99), 7.487 (1.72), 7.490 (1.60), 7.497 (4.92), 7.502 (4.85), 7.514 (12.36), 7.519 (1 1.80), 7.524 (10.39), 7.535 (1.82), 7.673 (0.57), 7.827 (3.06), 7.831 (2.83), 7.845 (4.22), 7.849 (4.10), 7.921 (8.1 1 ), 7.939 (5.57), 8.485 (1.76), 8.497 (2.96), 8.510 (1.72).
Beispiel 123
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(5-fluor-2-methoxyphenyl)- butansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 3-(5-fluor-2-methoxyphenyl)butanoat (100 mg, 165 μηηοΙ, Beispiel 207A) in Dichlormethan (1.5 ml) wurde TFA (250 μΙ, 3.3 mmol) gegeben, und das Gemisch wurde 16 h bei RT gerührt. An- schließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 20) gereinigt. Es wurden 85 mg (98% Reinheit, 92% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.01 min; MS (ESIpos): m/z = 551/553 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (1.91 ), 2.073 (0.62), 2.130 (10.47), 2.328 (0.42), 2.598 (0.87), 2.616 (0.89), 2.637 (3.99), 2.655 (6.87), 2.669 (4.44), 2.693 (0.99), 2.710 (1.01 ), 3.629 (0.89), 3.641 (1.93), 3.654 (1.67), 3.671 (2.33), 3.684 (1.55), 3.716 (0.77), 3.735 (1.75), 3.753 (2.34), 3.802 (2.87), 3.817 (1.90), 3.833 (1.57), 3.853 (0.85), 4.528 (0.50), 6.972 (1.13), 6.985 (1.67), 6.995 (5.05), 7.008 (6.28), 7.024 (2.40), 7.031 (2.66), 7.046 (0.67), 7.053 (0.86), 7.088 (2.87), 7.094 (2.60), 7.112 (2.95), 7.1 19 (2.50), 7.309 (0.53), 7.479 (0.71 ), 7.491 (1.84), 7.494 (1.88), 7.504 (6.38), 7.51 1 (5.58), 7.523 (16.00), 7.528 (15.38), 7.536 (9.41 ), 7.638 (1.59), 7.835 (3.16), 7.840 (2.92), 7.857 (4.89), 7.862 (4.72), 7.931 (8.83), 7.953 (5.66), 8.760 (1.91 ), 8.774 (3.65), 8.788 (1.89).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.77 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.64 (br. s, 1 H), 7.56-7.47 (m, 5H), 7.10 (dd, 1 H), 7.07-6.95 (m, 2H), 3.86-3.71 (m, 2H), 3.79 (s, 3H), 3.70- 3.60 (m, 1 H), 2.73-2.58 (m, 2H), 2.13 (br. s, 3H).
Beispiel 124
(+)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(5-fluor-2-methoxyphenyl)- butansäure (Enantiomer 1)
Zu einer Lösung aus (+)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3- (5-fluor-2-methoxyphenyl)butanoat (165 mg, 98% Reinheit, 266 μηηοΙ, Beispiel 209A) in Dichlormethan (1.9 ml) wurde TFA (410 μΙ, 5.3 mmol) gegeben und das Gemisch wurde 7 h bei RT gerührt.
Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 16) gereinigt. Es wurden 134 mg (98% Reinheit, 90% d. Th., ee-Wert 99%) der Titelverbindung erhalten.
[a]D 20 = +17.7°, 589 nm, c = 0.42 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.89 min; MS (ESIpos): m/z = 551/553 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (0.86), 2.073 (0.59), 2.129 (10.14), 2.327 (0.46), 2.519 (2.04), 2.524 (1.96), 2.597 (0.90), 2.615 (1.03), 2.636 (4.25), 2.654 (6.89), 2.669 (4.26), 2.692 (0.91 ), 2.709 (0.99), 3.628 (1.02), 3.640 (2.04), 3.653 (1.75), 3.670 (2.36), 3.684 (1.54), 3.716 (0.91 ), 3.734 (2.04), 3.752 (2.78), 3.801 (2.61 ), 3.816 (1.75), 3.833 (1.43), 3.853 (0.73), 6.972 (1.39), 6.985 (2.06), 6.995 (5.39), 7.008 (6.23), 7.023 (2.42), 7.031 (2.57), 7.046 (0.70), 7.053 (0.91 ), 7.087 (3.04), 7.094 (2.61 ), 7.1 1 1 (3.00), 7.1 18 (2.42), 7.478 (1.26), 7.489 (2.54), 7.493 (2.59), 7.503 (7.04), 7.51 1 (6.33), 7.522 (16.00), 7.527 (14.82), 7.535 (8.46), 7.539 (6.67), 7.547 (1.77), 7.638 (1.44), 7.833 (3.28), 7.838 (2.91 ), 7.856 (4.92), 7.861 (4.52), 7.930 (8.60), 7.952 (5.46), 8.757 (2.10), 8.772 (3.68), 8.785 (1.80).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.77 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.64 (br. s, 1 H), 7.57-7.44 (m, 5H), 7.10 (dd, 1 H), 7.07-6.93 (m, 2H), 3.88-3.70 (m, 2H), 3.78 (s, 3H), 3.66 (td, 1 H), 2.73-2.57 (m, 2H), 2.13 (br. s, 3H).
Beispiel 125
(-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(5-fluor-2-methoxyphenyl)- butansäure (Enantiomer 2)
Zu einer Lösung aus (-)-ierf-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(5- fluor-2-methoxyphenyl)butanoat (160 mg, 98% Reinheit, 258 pmol, Beispiel 208A) in Dichlormethan (1.8 ml) wurde TFA (400 μΙ, 5.2 mmol) gegeben und das Gemisch wurde 5 h bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan ver- setzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 16) gereinigt. Es wurden 1 16 mg (98% Reinheit, 80% d. Th., ee-Wert 99%) der Titelverbindung erhalten.
[a]D 20 = -17.5°, 589 nm, c = 0.36 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.90 min; MS (ESIpos): m/z = 551/553 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.05), 0.008 (0.98), 2.130 (10.36), 2.523 (1.05), 2.598 (0.88), 2.616 (0.89), 2.637 (4.20), 2.655 (7.04), 2.669 (4.51 ), 2.693 (1.03), 2.709 (1.06), 3.629 (0.86), 3.641 (1.97), 3.654 (1.64), 3.658 (1.64), 3.671 (2.35), 3.685 (1.54), 3.716 (0.68), 3.735 (1.69), 3.753 (2.26), 3.768 (2.21 ), 3.802 (2.78), 3.817 (1.83), 3.822 (1.40), 3.833 (1.51 ), 3.853 (0.80), 6.972 (1.20), 6.985 (1.79), 6.995 (5.46), 7.008 (6.55), 7.023 (2.45), 7.031 (2.75), 7.046 (0.67), 7.053 (0.91 ), 7.088 (3.00), 7.095 (2.67), 7.1 12 (3.09), 7.1 19 (2.62), 7.477 (0.74),
7.489 (1.89), 7.492 (1.85), 7.502 (6.44), 7.51 1 (5.82), 7.522 (16.00), 7.527 (15.40), 7.531 (12.19), 7.535 (9.42), 7.539 (7.62), 7.547 (2.13), 7.637 (1.48), 7.833 (3.40), 7.838 (3.1 1 ), 7.855 (5.24), 7.860 (5.04), 7.930 (9.35), 7.952 (6.05), 8.758 (1.97), 8.772 (3.76), 8.786 (1.91 ).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.77 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.64 (br. s, 1 H), 7.56-7.46 (m, 5H), 7.10 (dd, 1 H), 7.06-6.95 (m, 2H), 3.88-3.70 (m, 2H), 3.79 (s, 3H), 3.70- 3.62 (m, 1 H), 2.72-2.59 (m, 2H), 2.13 (br. s, 3H).
Beispiel 126
(+/.)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amin
butansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 3-(2-fluor-6-methoxyphenyl)butanoat (60 mg, 98.8 μηηοΙ, Beispiel 210A) in Dichlormethan (3.0 ml) wurde TFA (610 μΙ, 7.9 mmol) gegeben, und das Gemisch wurde 2.5 h bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlorme- than versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 15) gereinigt. Es wurden 45 mg (100% Reinheit, 83% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.88 min; MS (ESIpos): m/z = 551/553 [M+H]+
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: -0.007 (0.41 ), 1.234 (0.69), 2.073 (0.73), 2.148 (5.32), 2.518 (0.50), 2.725 (5.08), 2.728 (5.40), 2.741 (7.14), 2.759 (0.52), 3.686 (1.01 ), 3.698 (1.78), 3.71 1 (2.24), 3.723 (2.60), 3.735 (1.48), 3.793 (1.56), 3.802 (3.24), 3.836 (1.69), 3.849 (1.18), 3.944 (0.75), 3.959 (1.78), 3.974 (2.07), 3.989 (1.39), 4.005 (0.54), 6.736 (2.56), 6.754 (3.94), 6.772 (2.67), 6.834 (4.16), 6.851 (4.48), 7.201 (0.98), 7.217 (2.10), 7.231 (2.01 ), 7.247 (0.83), 7.473 (0.54), 7.476 (0.65), 7.481 (1.15), 7.490 (2.67), 7.492 (2.31 ), 7.495 (2.26), 7.502 (5.32), 7.504 (5.21 ), 7.507 (6.24), 7.520 (14.64), 7.526 (16.00), 7.531 (15.46), 7.542 (2.48), 7.683 (0.53), 7.837 (3.88), 7.842 (3.49), 7.855 (5.39), 7.860 (5.01 ), 7.929 (10.24), 7.947 (6.90), 8.818 (2.17), 8.830 (4.08), 8.842 (2.08).
1H-NMR (500 MHz, DMSO-d6): δ [ppm] = 12.05 (br. s, 1 H), 8.83 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.68 (br. s, 1 H), 7.55-7.46 (m, 5H), 7.27-7.17 (m, 1 H), 6.84 (d, 1 H), 6.75 (t, 1 H), 4.02-3.92 (m, 1 H), 3.87-3.77 (m, 1 H), 3.82 (s, 3H), 3.75-3.65 (m, 1 H), 2.76-2.70 (m, 2H), 2.15 (br. s, 3H).
Beispiel 127
(+)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-fl
butansäure (Enantiomer 1)
Zu einer Lösung aus (+)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3- (2-fluor-6-methoxyphenyl)butanoat (160 mg, 263 μηηοΙ, Beispiel 21 1A) in Dichlormethan (8.0 ml) wurde TFA (1.6 ml, 21 mmol) gegeben und das Gemisch wurde 24 h bei RT stehen gelassen. An- schließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 15) gereinigt. Es wurden 138 mg (100% Reinheit, 95% d. Th., ee-Wert 99%) der Titelverbindung erhalten.
[a]D 20 = +45.6°, 546 nm, c = 0.39 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.89 min; MS (ESIpos): m/z = 551/553 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 2.144 (5.94), 2.305 (0.40), 2.721 (5.05), 2.739 (6.25), 3.635 (1.06), 3.674 (1.38), 3.689 (1.90), 3.706 (2.26), 3.722 (2.51 ), 3.736 (1.55), 3.782 (1.28), 3.853 (0.99), 3.932 (0.61 ), 3.951 (1.44), 3.970 (1.71 ), 3.988 (1.13), 6.730 (1.94), 6.752 (3.05), 6.775 (2.1 1 ), 6.832 (3.38), 6.853 (3.76), 7.195 (0.82), 7.215 (1.83), 7.233 (1.81 ), 7.253 (0.71 ), 7.477 (0.57), 7.491 (1.63), 7.500 (4.91 ), 7.508 (4.59), 7.520 (16.00), 7.532 (7.41 ), 7.701 (0.51 ), 7.834 (2.47), 7.839 (2.22), 7.857 (3.87), 7.862 (3.65), 7.926 (6.94), 7.949 (4.31 ), 8.81 1 (1.60), 8.826 (3.15), 8.841 (1.57).
1 H-NMR (400 MHz, DMSO-d6): δ [ppm] = 12.04 (br. s, 1 H), 8.83 (t, 1 H), 7.93 (d, 1 H), 7.85 (dd, 1 H), 7.70 (br. s, 1 H), 7.56-7.46 (m, 5H), 7.22 (dd, 1 H), 6.84 (d, 1 H), 6.75 (t, 1 H), 4.03-3.91 (m, 1 H), 3.82 (s, 3H), 3.87-3.65 (m, 2H), 2.73 (d, 2H), 2.14 (br. s, 3H). Beispiel 128
(-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-fluor-6-methoxyphenyl)- butansäure (Enantiomer 2)
Zu einer Lösung aus (-)-ierf-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2- fluor-6-methoxyphenyl)butanoat (180 mg, 296 μηηοΙ, Beispiel 212A) in Dichlormethan (9.0 ml) wurde TFA (1.8 ml, 24 mmol) gegeben und das Gemisch wurde 24 h bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 15) gereinigt. Es wurden 163 mg (100% Reinheit, 100% d. Th., ee-Wert 99%) der Titelverbindung erhalten.
[a]D 20 = -39.2°, 546 nm, c = 0.39 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.89 min; MS (ESIpos): m/z = 551/553 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 2.144 (3.40), 2.720 (2.86), 2.739 (3.55), 3.674 (0.45), 3.689 (0.80), 3.706 (1.06), 3.722 (1.25), 3.736 (0.74), 3.781 (0.70), 3.801 (1.84), 3.818 (16.00), 3.853 (0.64), 3.932 (0.53), 3.950 (1.04), 3.969 (1.21 ), 3.988 (0.90), 4.007 (0.49), 6.730 (1.10), 6.752 (1.70), 6.775 (1.18), 6.832 (1.88), 6.853 (2.09), 7.195 (0.50), 7.215 (1.10), 7.234 (1.13), 7.253 (0.48), 7.491 (0.90), 7.501 (2.80), 7.509 (2.56), 7.521 (9.47), 7.835 (1.37), 7.840 (1.20), 7.858 (2.18), 7.863 (2.00), 7.927 (3.94), 7.949 (2.44), 8.812 (0.91 ), 8.827 (1.79), 8.841 (0.89).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 12.12 (br. s, 1 H), 8.83 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.69 (br. s, 1 H), 7.57-7.46 (m, 5H), 7.22 (dd, 1 H), 6.84 (d, 1 H), 6.75 (t, 1 H), 4.02-3.92 (m, 1 H), 3.82 (s, 3H), 3.87-3.66 (m, 2H), 2.73 (d, 2H), 2.14 (br. s, 3H).
Beispiel 129
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-ethoxyphenyl)butansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 3-(2-ethoxyphenyl)butanoat (40 mg, 98% Reinheit, 64.9 μηηοΙ, Beispiel 213A) in Dichlormethan (460 μΙ) wurde TFA (100 μΙ, 1 .3 mmol) gegeben, und das Gemisch wurde 3 Tage bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 16) gereinigt. Es wurden 32 mg (98% Reinheit, 88% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.00 min; MS (ESIpos): m/z = 547/549 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.15), 0.008 (1.15), 1.357 (7.81 ), 1.374 (16.00), 1.392 (7.54), 2.131 (7.94), 2.327 (0.46), 2.366 (0.53), 2.523 (2.32), 2.681 (6.13), 2.699 (6.24), 3.612 (0.81 ), 3.626 (1.44), 3.641 (1.57), 3.658 (1.87), 3.669 (2.00), 3.682 (1.94), 3.700 (2.04), 3.716 (1.35), 3.733 (0.46), 3.838 (1.13), 3.856 (1.63), 3.869 (1.52), 3.885 (1.39), 3.902 (0.63),
4.000 (0.43), 4.017 (1.07), 4.024 (1.46), 4.035 (1.99), 4.041 (3.68), 4.050 (3.25), 4.058 (3.67), 4.068 (2.95), 4.075 (1.71 ), 4.085 (1.07), 4.584 (0.53), 6.870 (1.82), 6.889 (3.70), 6.906 (2.12), 6.955 (3.54), 6.975 (4.16), 7.166 (1.80), 7.169 (2.03), 7.188 (2.95), 7.209 (4.78), 7.228 (3.16), 7.476 (0.97), 7.488 (1.95), 7.491 (2.08), 7.501 (5.51 ), 7.509 (5.16), 7.521 (13.33), 7.525 (12.65), 7.529 (9.54), 7.533 (7.34), 7.537 (5.94), 7.544 (1.76), 7.696 (1.94), 7.832 (2.65), 7.838 (2.40),
7.855 (3.99), 7.860 (3.78), 7.928 (7.02), 7.950 (4.45), 8.731 (1.63), 8.745 (2.66), 8.759 (1.55).
Beispiel 130
(-)-4-{[(6-Brom-3-methyl-2-phenyl^
(Enantiomer 1)
Zu einer Lösung aus (-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3- (2-ethoxyphenyl)butanoat (60 mg, 98% Reinheit, 97.4 μηηοΙ, Beispiel 214A) in Dichlormethan (690 μΙ) wurde TFA (150 μΙ, 1.9 mmol) gegeben und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde erneut TFA (150 μΙ, 1.9 mmol) hinzugegeben, und das Gemisch wurde weitere 4 h bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals hin- tereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels prä- parativer HPLC (Methode 16) gereinigt. Es wurden 51 mg (98% Reinheit, 94% d. Th., ee-Wert 99%) der Titelverbindung erhalten.
[a]D 20 = -14.2°, 589 nm, c = 0.36 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.99 min; MS (ESIpos): m/z = 547/549 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.357 (7.63), 1.374 (16.00), 1.391 (7.66), 2.131 (8.54), 2.328 (0.54), 2.681 (6.29), 2.699 (6.56), 3.613 (0.64), 3.627 (1.35), 3.641 (1.48), 3.658 (1.79), 3.669 (2.00), 3.682 (1.92), 3.700 (2.09), 3.716 (1.39), 3.734 (0.44), 3.838 (1.12), 3.856 (1.61 ), 3.869 (1.48), 3.886 (1.40), 3.902 (0.57), 4.024 (1.19), 4.041 (3.49), 4.050 (3.1 1 ), 4.058 (3.57), 4.067 (2.94), 4.085 (1.01 ), 6.869 (1.84), 6.888 (4.01 ), 6.906 (2.25), 6.955 (3.76), 6.975 (4.49), 7.169 (1.94), 7.188 (3.05), 7.209 (4.91 ), 7.228 (3.30), 7.477 (0.56), 7.492 (1.58), 7.502 (5.44), 7.509 (4.75), 7.521 (13.88), 7.525 (13.53), 7.533 (7.90), 7.696 (2.14), 7.834 (2.65), 7.839 (2.42),
7.856 (4.12), 7.861 (3.96), 7.928 (7.59), 7.951 (4.75), 8.732 (1.55), 8.746 (2.79), 8.760 (1.62).
Beispiel 131
(+)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-ethoxyphenyl)butansäure (Enantiomer 2)
Zu einer Lösung aus (+)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3- (2-ethoxyphenyl)butanoat (50 mg, 98% Reinheit, 81.2 μηηοΙ, Beispiel 215A) in Dichlormethan (580 μΙ) wurde TFA (130 μΙ, 1.6 mmol) gegeben und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde erneut TFA (150 μΙ, 1.9 mmol) hinzugegeben, und das Gemisch
wurde weitere 4 h bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 16) gereinigt. Es wurden 43 mg (100% Reinheit, 97% d. Th., ee- Wert 96%) der Titelverbindung erhalten. [a]D 20 = +15.0°, 546 nm, c = 0.37 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.99 min; MS (ESIpos): m/z = 547/549 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (1.19), 1.357 (7.49), 1.374 (16.00), 1.392 (7.67), 2.132 (7.97), 2.524 (1.23), 2.682 (6.05), 2.699 (6.38), 3.613 (0.64), 3.627 (1.28), 3.641 (1.40), 3.659 (1.71 ), 3.669 (1.87), 3.683 (1.84), 3.700 (2.00), 3.716 (1.30), 3.734 (0.42), 3.838 (0.99), 3.856 (1.53), 3.870 (1.44), 3.886 (1.34), 3.903 (0.56), 4.018 (0.73), 4.024 (1.13), 4.035 (1.63), 4.042 (3.43), 4.051 (3.00), 4.059 (3.54), 4.068 (2.85), 4.075 (1.64), 4.085 (0.97), 6.870 (1.76), 6.889 (3.86), 6.907 (2.19), 6.956 (3.66), 6.976 (4.33), 7.170 (1.87), 7.189 (2.88), 7.209 (4.73), 7.228 (3.18), 7.477 (0.46), 7.492 (1.38), 7.502 (4.96), 7.510 (4.35), 7.522 (12.96), 7.526 (12.44), 7.534 (7.29), 7.537 (6.06), 7.545 (1.75), 7.697 (1.92), 7.834 (2.42), 7.839 (2.25), 7.856 (3.82), 7.861 (3.77), 7.929 (7.09), 7.951 (4.49), 8.733 (1.45), 8.747 (2.65), 8.760 (1.58).
Beispiel 132
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(5-fluor-2-methoxyphenyl)-3- methylbutansäure (Racemat)
Zu einer Lösung aus (+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3- (5-fluor-2-methoxyphenyl)-3-methylbutanoat (80 mg, 98% Reinheit, 126 μηιοΙ, Beispiel 216A) in Dichlormethan (890 μΙ) wurde TFA (190 μΙ, 2.5 mmol) gegeben, und das Gemisch wurde 3 h bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Me- thode 16) gereinigt. Es wurden 63 mg (98% Reinheit, 87% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.96 min; MS (ESIpos): m/z = 565/567 [M+H]+
1H-NMR (500 MHz, DMSO-d6) δ [ppm]: 1.545 (16.00), 2.072 (1.45), 2.630 (4.20), 2.660 (4.55), 3.1 18 (3.18), 3.148 (2.85), 3.645 (1.16), 3.662 (1.21 ), 3.787 (0.43), 3.991 (0.88), 4.262 (1.15), 6.980 (2.89), 6.985 (3.88), 7.001 (4.1 1 ), 7.006 (5.59), 7.025 (1.87), 7.480 (0.98), 7.489 (2.17), 7.502 (4.86), 7.506 (5.01 ), 7.519 (1 1.64), 7.525 (10.85), 7.529 (10.71 ), 7.832 (2.53), 7.836 (2.44), 7.850 (3.51 ), 7.854 (3.44), 7.928 (6.72), 7.946 (4.63), 8.529 (1.69), 8.542 (2.64), 8.554 (1.59).
1H-NMR (500 MHz, DMSO-d6): δ [ppm] = 1 1.80 (br. s, 1 H), 8.54 (t, 1 H), 7.94 (d, 1 H), 7.84 (dd, 1 H), 7.66 (br. s, 1 H), 7.56-7.46 (m, 5H), 7.06-6.94 (m, 3H), 4.33-4.21 (m, 1 H), 3.85 (s, 3H), 3.69- 3.59 (m, 1 H), 3.13 (d, 1 H), 2.64 (d, 1 H), 2.09 (br. s, 3H), 1.55 (s, 3H). Beispiel 133
(+)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(5-fluor-2-meth
methylbutansäure (Enantiomer 1)
Zu einer Lösung aus (+)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3- (5-fluor-2-methoxyphenyl)-3-methylbutanoat (350 mg, 98% Reinheit, 552 μηηοΙ, Beispiel 217A) in Dichlormethan (3.9 ml) wurde TFA (850 μΙ, 1 1 mmol) gegeben und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 16) gereinigt. Es wurden 256 mg (98% Reinheit, 80% d. Th., ee-Wert 98%) der Titelverbindung erhalten. [a]D 20 = +14.2°, 589 nm, c = 0.35 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.04 min; MS (ESIpos): m/z = 565/567 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.29), 0.008 (1.82), 1.544 (16.00), 2.073 (1.83), 2.288 (0.43), 2.523 (1.02), 2.624 (4.25), 2.661 (4.92), 3.1 13 (3.61 ), 3.151 (3.22), 3.642 (2.21 ), 3.664 (2.55), 4.260 (1.06), 4.274 (1.01 ), 6.981 (3.74), 7.008 (7.08), 7.025 (2.13), 7.475 (0.68), 7.487 (1.66), 7.490 (1.82), 7.499 (5.60), 7.508 (5.27), 7.519 (15.60), 7.531 (7.16), 7.643 (0.46), 7.828 (2.57), 7.833 (2.30), 7.850 (3.95), 7.855 (3.70), 7.925 (7.40), 7.947 (4.69), 8.525 (1.67), 8.540 (2.68), 8.555 (1.57).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 1 1.84 (br. s, 1 H), 8.54 (t, 1 H), 7.93 (d, 1 H), 7.84 (dd, 1 H), 7.64 (br. s, 1 H), 7.56-7.45 (m, 5H), 7.07-6.94 (m, 3H), 4.27 (br. dd, 1 H), 3.85 (s, 3H), 3.65 (br. dd, 2H), 3.13 (d, 1 H), 2.64 (d, 1 H), 2.09 (br. s, 3H), 1.54 (s, 3H).
Beispiel 134
(-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(5-fluor-2-methoxyphenyl)-3- methylbutansäure (Enantiomer 2)
Zu einer Lösung aus (-)-ie f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3- (5-fluor-2-methoxyphenyl)-3-methylbutanoat (250 mg, 98% Reinheit, 394 μηιοΙ, Beispiel 218A) in Dichlormethan (2.8 ml) wurde TFA (610 μΙ, 7.9 mmol) gegeben und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinan- der mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 16) gereinigt. Es wurden 181 mg (98% Reinheit, 80% d. Th., ee-Wert 98%) der Titelverbindung erhalten.
[a]D 20 = -28.5°, 589 nm, c = 0.35 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.04 min; MS (ESIpos): m/z = 565/567 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.544 (15.55), 2.073 (2.25), 2.327 (0.51 ), 2.624 (3.82), 2.661 (4.55), 3.1 13 (3.45), 3.151 (3.03), 3.641 (1.26), 3.665 (1.41 ), 3.787 (0.56), 4.028 (0.86), 4.259 (1.46), 6.981 (3.61 ), 7.008 (6.97), 7.024 (2.23), 7.500 (5.47), 7.508 (5.24), 7.519 (16.00), 7.639 (0.53), 7.829 (2.44), 7.834 (2.28), 7.851 (3.80), 7.856 (3.62), 7.925 (6.86), 7.948 (4.33), 8.525 (1.60), 8.541 (2.69), 8.556 (1.58).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 1 1.86 (br. s, 1 H), 8.54 (t, 1 H), 7.93 (d, 1 H), 7.84 (dd, 1 H), 7.64 (br. s, 1 H), 7.56-7.46 (m, 5H), 7.06-6.96 (m, 3H), 4.31-4.22 (m, 1 H), 3.85 (s, 3H), 3.70- 3.60 (m, 1 H), 3.13 (d, 1 H), 2.64 (d, 1 H), 2.09 (br. s, 3H), 1.54 (s, 3H).
Beispiel 135
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(3,5-difluor-2-methoxy- phenyl)-3-methylbutansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 3-(3,5-difluor-2-methoxyphenyl)-3-methylbutanoat (80 mg, 98% Reinheit, 123 μηηοΙ, Beispiel 219A) in Dichlormethan (870 μΙ) wurde TFA (190 μΙ, 2.5 mmol) gegeben, und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels prä-
parativer HPLC (Methode 16) gereinigt. Es wurden 57 mg (98% Reinheit, 79% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.09 min; MS (ESIpos): m/z = 583/585 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (3.94), 0.008 (2.23), 1.159 (0.80), 1.175 (0.84), 1.194 (1.22), 1.205 (0.77), 1.21 1 (1.25), 1.222 (0.63), 1.433 (1.93), 1.546 (12.64), 2.073 (0.51 ), 2.159 (1.37), 2.327 (0.41 ), 2.518 (1.94), 2.523 (1.75), 2.637 (3.40), 2.675 (4.00), 3.121 (2.77), 3.159 (2.42), 3.663 (1.46), 3.677 (1.53), 3.696 (1.70), 3.710 (1.52), 3.964 (16.00), 3.969 (15.16), 4.055 (1.34), 4.072 (1.44), 4.087 (1.26), 4.105 (1.1 1 ), 6.885 (1.74), 6.91 1 (1.64), 7.188 (0.80), 7.194 (0.85), 7.215 (1.30), 7.237 (0.78), 7.480 (0.73), 7.491 (2.15), 7.506 (5.60), 7.512 (4.41 ), 7.524 (8.54), 7.537 (10.22), 7.556 (1.87), 7.840 (2.15), 7.846 (1.92), 7.863 (3.18), 7.868 (2.92), 7.938 (6.09), 7.961 (3.88), 8.619 (1.48), 8.634 (2.53), 8.649 (1.32).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 1 1.99 (br. s, 1 H), 8.63 (t, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.63 (br. s, 1 H), 7.57-7.45 (m, 5H), 7.27-7.14 (m, 1 H), 6.93-6.86 (m, 1 H), 4.08 (dd, 1 H), 3.97 (d, 3H), 3.69 (dd, 1 H), 3.14 (d, 1 H), 2.66 (d, 1 H), 2.16 (br. s, 3H), 1.55 (s, 3H). Beispiel 136
(+)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(3,5-difluor-2-methoxyphenyl)- 3-methylbutansäure (Enantiomer 1)
Zu einer Lösung aus (+)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3- (3,5-difluor-2-methoxyphenyl)-3-methylbutanoat (225 mg, 98% Reinheit, 345 μηηοΙ, Beispiel 220A) in Dichlormethan (2.4 ml) wurde TFA (530 μΙ, 6.9 mmol) gegeben und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels prä- parativer HPLC (Methode 16) gereinigt. Es wurden 158 mg (98% Reinheit, 77% d. Th., ee-Wert 93%) der Titelverbindung erhalten. [a]D 20 = +28.7°, 589 nm, c = 0.35 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.05 min; MS (ESIpos): m/z = 583/585 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.74), 0.007 (1.33), 1.547 (13.49), 2.073 (1.33), 2.159 (1.39), 2.637 (3.51 ), 2.676 (4.10), 3.121 (2.91 ), 3.159 (2.57), 3.663 (1.56), 3.677 (1.62), 3.696 (1.81 ), 3.710 (1.65), 3.964 (16.00), 3.969 (15.76), 4.055 (1.36), 4.072 (1.48), 4.088 (1.30), 4.105 (1.18), 6.885 (1.76), 6.912 (1.74), 7.193 (0.83), 7.215 (1.36), 7.237 (0.81 ), 7.479 (0.52), 7.492 (1.78), 7.506 (5.43), 7.512 (4.18), 7.524 (8.81 ), 7.537 (10.58), 7.555 (2.06), 7.841 (2.07), 7.846 (1.91 ), 7.863 (3.18), 7.868 (3.05), 7.939 (6.26), 7.961 (4.00), 8.619 (1.42), 8.635 (2.66), 8.650 (1.40).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 12.00 (br. s, 1 H), 8.63 (t, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.68 (br. s, 1 H), 7.57-7.46 (m, 5H), 7.27-7.17 (m, 1 H), 6.90 (d, 1 H), 4.08 (dd, 1 H), 3.97 (d, 3H), 3.69 (dd, 1 H), 3.14 (d, 1 H), 2.66 (d, 1 H), 2.16 (br. s, 3H), 1.55 (s, 3H).
Beispiel 137
(-)-4-{[(6-Brom-3-methyl-2-phenylchi^
3-methylbutansäure (Enantiomer 2)
Zu einer Lösung aus (-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3- (3,5-difluor-2-methoxyphenyl)-3-methylbutanoat (225 mg, 98% Reinheit, 345 μιτιοΙ, Beispiel 221A) in Dichlormethan (2.4 ml) wurde TFA (530 μΙ, 6.9 mmol) gegeben und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 16) gereinigt. Es wurden 159 mg (98% Reinheit, 78% d. Th., ee-Wert 98%) der Titelverbindung erhalten.
[a]D 20 = -31.6°, 589 nm, c = 0.45 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.05 min; MS (ESIpos): m/z = 583/585 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.48), 1.547 (12.71 ), 2.073 (0.80), 2.160 (1.28), 2.519 (0.84), 2.524 (0.68), 2.638 (3.41 ), 2.676 (3.97), 3.122 (2.77), 3.160 (2.46), 3.663 (1.63), 3.677 (1.66), 3.696 (1.80), 3.71 1 (1.64), 3.965 (16.00), 3.970 (15.57), 4.056 (1.31 ), 4.073 (1.41 ), 4.089 (1.25), 4.106 (1.13), 6.886 (1.66), 6.913 (1.63), 7.187 (0.69), 7.194 (0.75), 7.216 (1.25), 7.237 (0.75), 7.244 (0.69), 7.480 (0.49), 7.492 (1.79), 7.507 (5.17), 7.513 (3.97), 7.525 (8.15), 7.538 (10.19), 7.557 (1.89), 7.841 (2.06), 7.847 (1.86), 7.864 (3.15), 7.869 (2.99), 7.940 (6.19), 7.962 (3.98), 8.620 (1.35), 8.636 (2.51 ), 8.651 (1.33).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 1 1.99 (br. s, 1 H), 8.64 (t, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.67 (br. s, 1 H), 7.57-7.46 (m, 5H), 7.26-7.17 (m, 1 H), 6.90 (d, 1 H), 4.08 (dd, 1 H), 3.97 (d, 3H), 3.69 (dd, 1 H), 3.14 (d, 1 H), 2.66 (d, 1 H), 2.16 (br. s, 3H), 1.55 (s, 3H).
Beispiel 138
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(4-fluor-2-methoxyphenyl)-3- methylbutansäure (Racemat)
Zu einer Lösung aus (+/-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3- (4-fluor-2-methoxyphenyl)-3-methylbutanoat (80 mg, 98% Reinheit, 126 mol, Beispiel 222A) in Dichlormethan (890 μΙ) wurde TFA (190 μΙ, 2.5 mmol) gegeben, und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 16) gereinigt. Es wurden 43 mg (98% Reinheit, 59% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.06 min; MS (ESIpo): m/z = 565/567 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.84), 0.008 (2.39), 1.549 (14.23), 2.092 (1.63), 2.286 (0.61 ), 2.327 (0.44), 2.523 (1.26), 2.594 (4.23), 2.631 (4.77), 2.669 (0.42), 2.853 (0.52), 3.060 (4.14), 3.097 (3.69), 3.603 (3.87), 3.617 (4.23), 3.636 (4.36), 3.649 (4.02), 4.229 (1.86), 4.246 (2.03), 4.261 (1.86), 4.279 (1.71 ), 6.666 (1.05), 6.672 (1.20), 6.687 (2.07), 6.693 (2.22), 6.708 (1.18), 6.714 (1.17), 6.857 (2.08), 6.863 (2.1 1 ), 6.886 (2.19), 6.891 (2.05), 7.192 (2.56), 7.209 (3.14), 7.213 (3.09), 7.231 (2.42), 7.474 (0.64), 7.488 (1.81 ), 7.498 (5.53), 7.506 (5.27), 7.516 (16.00), 7.528 (7.27), 7.532 (6.20), 7.618 (0.43), 7.645 (0.44), 7.825 (2.60), 7.831 (2.39), 7.848 (4.02), 7.853 (3.90), 7.920 (7.70), 7.943 (4.84), 8.497 (1.71 ), 8.513 (2.89), 8.528 (1.62).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 1 1.81 (br. s, 1 H), 8.51 (t, 1 H), 7.93 (d, 1 H), 7.84 (dd, 1 H), 7.66 (br. s, 1 H), 7.56-7.45 (m, 5H), 7.21 (dd, 1 H), 6.87 (dd, 1 H), 6.69 (td, 1 H), 4.25 (dd, 1 H), 3.86 (s, 3H), 3.63 (dd, 1 H, teilweise verdeckt), 3.08 (d, 1 H), 2.61 (d, 1 H), 2.09 (br. s, 3H), 1.55 (s, 3H).
Beispiel 139
(+)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(4-fluor-2-methoxyphenyl)-3- methylbutansäure (Enantiomer 1)
Zu einer Lösung aus (+)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3- (4-fluor-2-methoxyphenyl)-3-methylbutanoat (650 mg, 98% Reinheit, 1.02 mmol, Beispiel 223A)
in Dichlormethan (7.3 ml) wurde TFA (1.6 ml, 20 mmol) gegeben und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 16) gereinigt. Es wurden 553 mg (98% Reinheit, 94% d. Th., ee-Wert 98%) der Titelverbindung erhalten.
[a]D 20 = +27.5°, 589 nm, c = 0.36 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.98 min; MS (ESIpos): m/z = 565/567 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.57), 0.008 (1.42), 1.550 (14.64), 2.030 (0.71 ), 2.072 (4.01 ), 2.097 (1.69), 2.286 (0.55), 2.523 (0.91 ), 2.595 (4.29), 2.632 (4.86), 2.854 (0.49), 3.061 (4.19), 3.097 (3.74), 3.604 (2.68), 3.617 (3.05), 3.636 (3.50), 3.650 (3.52), 3.680 (2.03), 4.230 (1.90), 4.247 (2.09), 4.262 (1.92), 4.280 (1.77), 6.666 (1.07), 6.672 (1.23), 6.688 (2.13), 6.693 (2.28), 6.708 (1.21 ), 6.714 (1.21 ), 6.858 (2.13), 6.863 (2.17), 6.886 (2.19), 6.891 (2.11 ), 7.192 (2.56), 7.210 (3.18), 7.214 (3.14), 7.231 (2.45), 7.475 (0.59), 7.488 (1.77), 7.498 (5.58), 7.506 (5.27), 7.517 (16.00), 7.529 (7.63), 7.613 (0.45), 7.648 (0.45), 7.826 (2.59), 7.831 (2.43), 7.849 (4.03), 7.854 (3.96), 7.921 (7.64), 7.944 (4.84), 8.498 (1.71 ), 8.513 (2.95), 8.529 (1.67).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 1 1.78 (br. s, 1 H), 8.51 (t, 1 H), 7.93 (d, 1 H), 7.84 (dd, 1 H), 7.65 (br. s, 1 H), 7.55-7.47 (m, 5H), 7.21 (dd, 1 H), 6.87 (dd, 1 H), 6.69 (td, 1 H), 4.25 (dd, 1 H), 3.86 (s, 3H), 3.63 (dd, 1 H, teilweise verdeckt), 3.08 (d, 1 H), 2.61 (d, 1 H), 2.10 (br. s, 3H), 1.55 (s, 3H). Beispiel 140
(-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(4-fluor-2-methoxyphenyl)-3- methylbutansäure (Enantiomer 2)
Zu einer Lösung aus (-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3- (4-fluor-2-methoxyphenyl)-3-methylbutanoat (680 mg, 98% Reinheit, 1.07 mmol, Beispiel 224A) in Dichlormethan (7.6 ml) wurde TFA (1.7 ml, 21 mmol) gegeben, und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 16) gereinigt. Es wurden 568 mg (98% Reinheit, 92% d. Th., ee-Wert 98%) der Titelverbindung erhalten. [a]D 20 = -27.0°, 589 nm, c = 0.46 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.98 min; MS (ESIpos): m/z = 565/567 [M+H]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (1.45), 1.549 (14.1 1 ), 2.073 (1.75), 2.094 (1.73), 2.287 (0.48), 2.523 (1.72), 2.595 (4.05), 2.632 (4.55), 2.854 (0.41 ), 3.061 (4.00), 3.097 (3.53), 3.604 (1.78), 3.617 (1.92), 3.636 (2.06), 3.650 (1.85), 4.043 (0.45), 4.230 (2.75), 4.248 (2.96),
4.263 (2.80), 4.280 (2.63), 6.672 (1.25), 6.688 (2.10), 6.693 (2.21 ), 6.708 (1.18), 6.714 (1.15), 6.858 (2.08), 6.863 (2.13), 6.886 (2.16), 6.891 (2.01 ), 7.192 (2.48), 7.210 (3.10), 7.213 (3.05), 7.231 (2.29), 7.477 (0.75), 7.489 (2.14), 7.500 (5.94), 7.508 (5.81 ), 7.518 (16.00), 7.618 (0.47), 7.646 (0.47), 7.828 (2.57), 7.833 (2.38), 7.851 (3.89), 7.856 (3.74), 7.923 (7.25), 7.945 (4.55), 8.499 (1.80), 8.515 (2.91 ), 8.530 (1.64).
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 1 1.78 (br. s, 1 H), 8.51 (t, 1 H), 7.93 (d, 1 H), 7.84 (dd, 1 H), 7.65 (br. s, 1 H), 7.56-7.46 (m, 5H), 7.21 (dd, 1 H), 6.87 (dd, 1 H), 6.69 (td, 1 H), 4.26 (dd, 1 H), 3.86 (s, 3H), 3.63 (dd, 1 H, teilweise verdeckt), 3.08 (d, 1 H), 2.61 (d, 1 H), 2.09 (br. s, 3H), 1.55 (s, 3H). Beispiel 141
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinoli^
phenyl)butansäure (Racemat)
Zu einer Lösung aus einem Gemisch von (+/-)-Ethyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4- yl)carbonyl]amino}-2,2-difluor-3-(2-methoxyphenyl)butanoat und (+/-)-Methyl-4-{[(6-brom-3-methyl- 2-phenylchinolin-4-yl)carbonyl]amino}-2,2-difluor-3-(2-methoxyphenyl)butanoat (40 mg, ca. 67 μη-ιοΙ, Beispiel 225A) in THF (1 ml) wurden Wasser (70 μΙ) und Lithiumhydroxid (3.2 mg, 134 μΜ) gegeben, und das Gemisch wurde 1 h bei RT gerührt. Anschließend wurde erneut Lithiumhydroxid (4.8 mg, 200 μΜ) hinzugegeben, und das Gemisch wurde eine weitere Stunde bei RT gerührt. Danach wurde das Gemisch mit 1 M Salzsäure (350 μΙ, 330 μηηοΙ) versetzt und mittels praparativer HPLC (Chromatorex C18, 10 μΜ, 125 x 30 mm, Acetonitril/Wasser-Gradient mit 0.01% TFA) vorgereinigt. Die vereinigten Zielfraktionen wurden eingeengt, der Rückstand lyophilisiert und erneut mittels praparativer HPLC (Chromatorex C18, 10 μΜ, 125 x 30 mm, Acetonitril/Wasser-Gradient mit 0.01 % TFA) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyo- philisiert Es wurden 13 mg (95% Reinheit, 31 % d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.77 min; MS (ESIpos): m/z = 569/571 [M+H]+
1H-NMR (600 MHz, DMSO-d6) δ [ppm]: 0.005 (1.53), 1.1 12 (0.69), 1.162 (0.52), 1.174 (0.86), 1.186 (0.42), 1.383 (0.49), 1.468 (1.69), 2.019 (1.58), 2.386 (0.55), 2.517 (1.58), 2.520 (1.52), 2.523 (1.23), 2.614 (0.58), 3.967 (2.73), 3.976 (1.98), 4.452 (0.53), 4.465 (0.65), 4.479 (0.96), 4.491 (0.98), 4.504 (0.95), 6.976 (0.78), 6.988 (1.48), 7.001 (0.92), 7.016 (2.53), 7.030 (2.72), 7.287 (0.81 ), 7.300 (1.34), 7.373 (2.30), 7.385 (2.12), 7.473 (0.59), 7.483 (2.04), 7.490 (3.04), 7.500 (15.67), 7.502 (16.00), 7.509 (3.34), 7.514 (2.23), 7.524 (0.92), 7.824 (1.91 ), 7.828 (1.84), 7.839 (2.64), 7.842 (2.58), 7.910 (5.19), 7.925 (3.69), 8.872 (1.42), 8.882 (2.72), 8.891 (1.42).
1H-NMR (600 MHz, DMSO-d6): δ [ppm] = 14.69 (br. s, 1 H), 8.88 (t, 1 H), 7.92 (d, 1 H), 7.83 (dd, 1 H), 7.63 (br. s, 1 H), 7.54-7.46 (m, 5H), 7.38 (d, 1 H), 7.30 (t, 1 H), 7.02 (d, 1 H), 6.99 (t, 1 H), 4.54- 4.45 (m, 1 H), 4.01-3.91 (m, 2H), 3.75 (s, 3H), 2.02 (br. s, 3H).
Beispiel 142
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-fluor-6^
methylbutansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 3-(2-fluor-6-methoxyphenyl)-3-methylbutanoat (40 mg, 84% Reinheit, 54 μηηοΙ, Beispiel 226A) in Dichlormethan (800 μΙ) wurde TFA (83 μΙ, 1.1 mmol) gegeben, und das Gemisch wurde über Nacht bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels prä- parativer HPLC (Methode 15) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 30 mg (90% Reinheit, 90 % d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.96 min; MS (ESIpos): m/z = 565/567 [M+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 1 1.67 (br. s, 1 H), 8.68 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.64 (br. s, 1 H), 7.56-7.46 (m, 5H), 7.26-7.14 (m, 1 H), 6.83 (d, 1 H), 6.67 (dd, 1 H), 4.17 (dd, 1 H), 3.83 (s, 3H), 3.67 (dd, 1 H), 3.33 (d, 1 H), 2.48 (d, 1 H, teilweise verdeckt), 2.12 (s, 3H), 1.69 (d, 3H).
Beispiel 143
(+)-4-{[(6-Brom-3-methyl-2-phenylch^
methylbutansäure (Enantiomer 1)
Zu einer Lösung aus (+)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3- (2-fluor-6-methoxyphenyl)-3-methylbutanoat (66 mg, 87% Reinheit, 91.8 μηηοΙ, Beispiel 228A) in Dichlormethan (1.4 ml) wurde TFA (140 μΙ, 1.8 mmol) gegeben und das Gemisch wurde 3 Tage bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präpara- tiver HPLC (Methode 15) gereinigt. Es wurden 56 mg (85% Reinheit, 92% d. Th., ee-Wert >99%) der Titelverbindung erhalten.
[a]D 20 = +13.8°, 589 nm, c = 0.39 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.07 min; MS (ESIpos): m/z = 565/567 [M+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.67 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.64 (br. s, 1 H), 7.56-7.47 (m, 5H), 7.25-7.15 (m, 1 H), 6.83 (d, 1 H), 6.67 (dd, 1 H), 4.17 (br. dd, 1 H), 3.83 (s, 3H), 3.67 (br. dd, 1 H), 3.33 (d, 1 H), 2.50 (d, 1 H, teilweise verdeckt), 2.12 (br. s, 3H), 1 .69 (d, 3H).
Beispiel 144
(-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-fluor-6-methoxyphenyl)-3- methylbutansäure (Enantiomer 2)
Zu einer Lösung aus (-)-ie/f-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3- (2-fluor-6-methoxyphenyl)-3-methylbutanoat (51 mg, 82.1 μηηοΙ, Beispiel 227A) in Dichlormethan (1.2 ml) wurde TFA (130 μΙ, 1.6 mmol) gegeben und das Gemisch wurde über Nacht bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 15) gereinigt. Es wurden 45 mg (100% Reinheit, 96% d. Th., ee-Wert 96%) der Titel- Verbindung erhalten.
[a]D 20 = -13.4°, 589 nm, c = 0.37 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.07 min; MS (ESIpos): m/z = 565/567 [M+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.68 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.64 (br. s, 1 H), 7.57-7.47 (m, 5H), 7.24-7.15 (m, 1 H), 6.83 (d, 1 H), 6.67 (dd, 1 H), 4.17 (dd, 1 H), 3.83 (s, 3H), 3.67 (dd, 1 H), 3.33 (d, 1 H), 2.50 (d, 1 H, teilweise verdeckt), 2.12 (br. s, 3H), 1.69 (d, 3H).
Beispiel 145
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-chlorphenyl)pentansäure (Racemat 1)
Zu einer Lösung aus (+/-)-tert-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3- (2-chlorphenyl)pentanoat (109 mg, 179 μηιοΙ, Beispiel 230A, Racemat 1 ) in Dichlormethan (4.1 ml) wurde TFA (410 μΙ, 5.4 mmol) gegeben, und das Gemisch wurde 16 h bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 17) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand in Aceton itril/Wasser lyophilisiert. Es wurden 82 mg (100% Reinheit, 83% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.98 min; MS (ESIpos): m/z = 551/553 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (1.20), 1.237 (13.23), 1.254 (13.14), 1.908 (0.66), 2.073 (0.72), 2.273 (1.13), 2.31 1 (0.99), 2.327 (0.85), 2.366 (0.56), 2.698 (1.30), 2.710 (0.55), 2.720 (1.29), 2.739 (3.98), 2.761 (4.71 ), 2.769 (4.90), 2.782 (4.83), 2.809 (1.55), 2.823 (1.22), 3.762 (1.29), 3.783 (2.61 ), 3.796 (2.76), 3.817 (1.23), 4.656 (1.22), 4.676 (2.24), 4.694 (2.21 ), 4.713 (1.15), 7.298 (1.38), 7.353 (2.08), 7.422 (2.40), 7.469 (0.98), 7.477 (1.37), 7.490 (3.23), 7.506 (9.56), 7.51 1 (1 1.16), 7.524 (16.00), 7.536 (13.89), 7.831 (2.32), 7.853 (3.43), 7.929 (5.38), 7.952 (3.55), 8.767 (1.35).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.06 (br. s, 1 H), 8.77 (br. s, 1 H), 7.94 (d, 1 H), 7.84 (d, 1 H), 7.67-7.15 (m, 10H), 4.78-4.54 (m, 1 H), 3.86-3.69 (m, 1 H), 2.87-2.63 (m, 2H), 2.44-1.69 (br. m, 3H), 1.25 (d, 3H).
Trennung der Enantiomere:
Die Titelverbindung (75 mg) wurde mittels präparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 146 und 147) [Säule: Daicel Chiralpak AD-H, 5 μηη, 250 mm x 20 mm; Fluss: 40 ml/min; Detektion: 220 nm; Injektion: 0.25 ml; Eluent: 70% Heptan / 30% Ethanol; isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand in Ac- tonitril/Wasser lyophilisiert.
Beispiel 146
(-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-chlorph
(Enantiomer 1)
Bei der in Beispiel 145 beschriebenen Enantiomerentrennung wurden 38 mg (100% Reinheit, ee-Wert 99%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -8.3°, 589 nm, c = 0.32 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.97 min; MS (ESIpos): m/z = 551/553 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.19), 0.008 (2.01 ), 1.240 (12.95), 1.256 (12.80), 1.897 (0.65), 2.274 (1.13), 2.312 (0.98), 2.328 (1.02), 2.366 (0.61 ), 2.670 (0.51 ), 2.707 (1.20), 2.728 (1.12), 2.747 (3.94), 2.772 (6.13), 2.786 (4.84), 2.813 (1 .43), 2.827 (1.08), 3.762 (1.30), 3.783 (2.58), 3.796 (2.77), 3.818 (1.28), 4.501 (1.14), 4.662 (2.03), 4.681 (2.98), 4.699 (2.91 ), 4.719 (1.83), 7.351 (2.03), 7.423 (2.35), 7.480 (1.28), 7.492 (2.99), 7.508 (9.17), 7.514 (10.79), 7.525 (16.00), 7.538 (13.75), 7.835 (2.31 ), 7.857 (3.40), 7.931 (5.43), 7.954 (3.52), 8.741 (1.31 ).
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.74 (br. s, 1 H), 7.94 (d, 1 H), 7.85 (d, 1 H), 7.77-7.13 (m, 10H), 4.76-4.62 (m, 1 H), 3.85-3.72 (m, 1 H), 2.88-2.64 (m, 2H), 2.39-1.74 (br. m, 3H), 1.25 (d, 3H).
Beispiel 147
(+)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-chlorphenyl)pentansäure (Enantiomer 2)
Bei der in Beispiel 145 beschriebenen Enantiomerentrennung wurden 38 mg (100% Reinheit, ee-Wert 99%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = +12.0°, 589 nm, c = 0.33 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.97 min; MS (ESIpos): m/z = 551/553 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.22), 0.008 (1.95), 1.239 (13.31 ), 1.256 (13.07), 1.901 (0.65), 2.272 (1.16), 2.31 1 (0.98), 2.328 (1.06), 2.366 (0.64), 2.670 (0.56), 2.706 (1.25), 2.728 (1.14), 2.747 (4.02), 2.772 (6.21 ), 2.786 (4.89), 2.813 (1.47), 2.826 (1.10), 3.762 (1.35), 3.783 (2.64), 3.796 (2.83), 3.817 (1.32), 4.370 (1.38), 4.661 (1.70), 4.681 (2.69), 4.698 (2.64), 4.719 (1.54), 7.352 (1.99), 7.422 (2.29), 7.480 (1.20), 7.492 (2.87), 7.508 (9.04), 7.514 (10.65), 7.525 (16.00), 7.538 (13.81 ), 7.835 (2.29), 7.857 (3.41 ), 7.931 (5.41 ), 7.953 (3.58), 8.743 (1.32). Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.74 (br. s, 1 H), 7.94 (d, 1 H), 7.85 (d, 1 H), 7.78-7.1 1 (m, 10H), 4.76-4.62 (m, 1 H), 3.86-3.69 (m, 1 H), 2.87-2.61 (m, 2H), 2.39-1.71 (br. m, 3H), 1.25 (d, 3H).
Beispiel 148
(+/-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amin
(Racemat 2)
Zu einer Lösung aus (+/-)-ierf-Butyl-4-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 3-(2-chlorphenyl)pentanoat (143 mg, 236 μmol, Beispiel 231A, Racemat 2, nicht reinheitskorri- giert) in Dichlormethan (5.4 ml) wurde TFA (550 μΙ, 7.1 mmol) gegeben, und das Gemisch wurde 16 h bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals hintereinander mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels prä- parativer HPLC (Methode 17) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand in Aceton itril/Wasser lyophilisiert. Es wurden 1 1 1 mg (100% Reinheit, 86% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.08 min; MS (ESIpos): m/z = 551/553 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (2.38), 1.120 (15.15), 1.136 (15.18), 2.287 (1 1.03), 2.366 (0.80), 2.523 (1.59), 2.670 (0.67), 2.710 (0.58), 2.794 (0.75), 2.835 (2.56), 2.863 (6.52), 2.875 (5.06), 2.903 (1.42), 2.915 (0.86), 3.755 (1.32), 3.774 (2.51 ), 3.791 (2.41 ), 3.812
(1.29) , 4.481 (2.28), 4.500 (2.25), 7.285 (2.60), 7.342 (2.37), 7.361 (3.86), 7.379 (2.19), 7.443 (7.20), 7.458 (8.84), 7.476 (4.56), 7.488 (1.67), 7.507 (4.27), 7.523 (16.00), 7.542 (14.10), 7.557 (5.25), 7.563 (3.96), 7.582 (13.25), 7.585 (13.78), 7.601 (7.42), 7.605 (6.1 1 ), 7.712 (1.08), 7.871
(5.30) , 7.876 (4.76), 7.893 (7.73), 7.898 (7.35), 7.976 (12.07), 7.998 (7.91 ), 8.918 (0.82), 12.022 (0.72).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.02 (br. s, 1 H), 8.92 (br. s, 1 H), 7.98 (d, 1 H), 7.89 (dd, 1 H), 7.71 (br. s, 1 H), 7.62-7.41 (m, 7H), 7.41-7.23 (m, 2H), 4.58-4.34 (m, 1 H), 3.90-3.69 (m, 1 H), 2.95-2.74 (m, 2H), 2.29 (br. s, 3H), 1.13 (d, 3H).
Trennung der Enantiomere:
Die Titelverbindung (100 mg) wurde in Ethanol (5 ml) gelöst und mittels präparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 149 und 150) [Säule: YMC Chiralart Amylose SA, 5 μηη, 250 mm x 30 mm; Fluss: 30 ml/min; Detektion: 220 nm; Temperatur: 50 °C; Injektion: 0.4 ml; Eluent: 50% Heptan / 50% Ethanol; Laufzeit 9 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand in Acton itril/Wasser lyophilisiert. Beispiel 149
(-)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-chlorphenyl)pentansäure (Enantiomer 3)
Bei der in Beispiel 148 beschriebenen Enantiomerentrennung wurden 51 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -43.1 °, 589 nm, c = 0.31 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.09 min; MS (ESIpos): m/z = 551/553 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.120 (15.75), 1.136 (16.00), 1.234 (1.04), 1.906 (5.70),
2.287 (1 1.95), 2.366 (0.59), 2.670 (0.51 ), 2.709 (0.50), 2.796 (0.79), 2.836 (2.62), 2.863 (6.36), 2.875 (5.02), 2.903 (1.40), 3.757 (1.40), 3.775 (2.72), 3.791 (2.60), 4.482 (2.49), 4.501 (2.48),
7.285 (2.88), 7.342 (2.62), 7.360 (4.13), 7.377 (2.37), 7.442 (7.42), 7.458 (9.1 1 ), 7.475 (5.06), 7.506 (4.47), 7.522 (15.99), 7.541 (14.31 ), 7.556 (5.31 ), 7.584 (14.44), 7.600 (7.51 ), 7.708 (1.13), 7.870 (4.75), 7.875 (4.52), 7.893 (6.97), 7.897 (6.98), 7.975 (1 1.38), 7.997 (7.40), 8.915 (0.92), 12.023 (0.72).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.02 (br. s, 1 H), 8.92 (br. s, 1 H), 7.98 (d, 1 H), 7.88 (dd, 1 H), 7.71 (br. s, 1 H), 7.62-7.42 (m, 7H), 7.41-7.23 (m, 2H), 4.56-4.43 (m, 1 H), 3.84-3.73 (m, 1 H), 2.94-2.75 (m, 2H), 2.29 (br. s, 3H), 1.13 (d, 3H).
Beispiel 150
(+)-4-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(2-chlorphenyl)pentansäure (Enantiomer 4)
Bei der in Beispiel 148 beschriebenen Enantiomerentrennung wurden 50 mg (100% Reinheit, ee-Wert 99%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = +44.8°, 589 nm, c = 0.34 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.09 min; MS (ESIpos): m/z = 551/553 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.121 (15.53), 1.137 (15.76), 1.234 (0.98), 1.91 1 (3.31 ),
2.288 (1 1.97), 2.366 (0.64), 2.670 (0.42), 2.710 (0.50), 2.805 (0.74), 2.845 (2.61 ), 2.869 (6.06), 2.908 (1.29), 3.779 (2.66), 3.795 (2.57), 4.487 (2.46), 4.505 (2.45), 7.287 (2.85), 7.344 (2.57), 7.362 (4.10), 7.379 (2.37), 7.444 (6.97), 7.459 (8.99), 7.476 (5.05), 7.488 (1.97), 7.507 (4.50), 7.523 (16.00), 7.543 (14.48), 7.557 (5.51 ), 7.563 (4.31 ), 7.583 (13.32), 7.585 (14.36), 7.601 (7.69), 7.605 (6.62), 7.707 (1.19), 7.871 (5.03), 7.876 (4.79), 7.894 (7.43), 7.899 (7.37), 7.976 (1 1.53), 7.999 (7.62), 8.896 (1.08), 12.013 (1.09).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.01 (br. s, 1 H), 8.90 (br. s, 1 H), 7.99 (d, 1 H), 7.89 (dd, 1 H), 7.71 (br. s, 1 H), 7.63-7.42 (m, 7H), 7.40-7.22 (m, 2H), 4.57-4.41 (m, 1 H), 3.85-3.72 (m, 1 H), 2.96-2.74 (m, 2H), 2.29 (br. s, 3H), 1.13 (d, 3H). Beispiel 151
(+/-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlorphenyl)pentansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 4-(2-chlorphenyl)pentanoat (1.40 g, 2.30 mmol, Beispiel 232A) in Dichlormethan (16 ml) wurde TFA (3.5 ml, 46 mmol) gegeben und das Gemisch wurde 7 h bei RT gerührt. Anschließend wur- de das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode (Methode 19) gereinigt. Es wurden 1.17 g (98% Reinheit, 90% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.97 min; MS (ESIpos): m/z = 551/553 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.75), 1.805 (0.47), 1.824 (0.99), 1.847 (1.30), 1.870 (0.70), 2.034 (0.64), 2.054 (2.58), 2.073 (10.29), 2.089 (2.87), 2.100 (2.84), 2.141 (4.96), 2.170 (1.18), 3.688 (1.28), 3.707 (1.72), 3.720 (2.05), 7.249 (0.90), 7.267 (1.95), 7.286 (1.35), 7.358 (1.18), 7.376 (2.1 1 ), 7.394 (1.10), 7.438 (3.36), 7.440 (3.41 ), 7.458 (2.79), 7.460 (2.78), 7.500 (6.51 ), 7.508 (4.47), 7.520 (16.00), 7.531 (5.82), 7.694 (0.48), 7.830 (2.13), 7.835 (1.98), 7.853 (3.18), 7.858 (3.14), 7.928 (5.99), 7.950 (3.77), 8.841 (1.29), 8.855 (2.54), 8.870 (1.23). 1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1 1.99 (br. s, 1 H), 8.86 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.69 (br. s, 1 H), 7.56-7.48 (m, 6H), 7.45 (dd, 1 H), 7.38 (br. t, 1 H), 7.27 (br. t, 1 H), 3.87-3.49 (m, 3H), 2.22-2.02 (m, 6H), 1.92-1.71 (m, 1 H).
Trennung der Enantiomere:
Die Titelverbindung (1.0 g) wurde in einem 1 :1-Gemisch aus Acetonitril und Methanol (100 ml) gelöst und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 152 und 153) [Säule: Daicel Chiralpak AD-H, 5 μηι, 250 mm x 30 mm; Fluss: 100 ml/min; Detektion: 210 nm; Temperatur: 38 °C; Injektion: 0.6 ml; Eluent: 68% Kohlendioxid / 32% Methanol; Laufzeit 14 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet.
Beispiel 152
(-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4^
(Enantiomer 1)
Bei der in Beispiel 151 beschriebenen Enantiomerentrennung wurden 518 mg (98% Reinheit, ee-Wert 98%) der Titelverbindung als früher eluierendes Enantiomer erhalten. Die Substanz wurde anschließend lyophilisiert.
[a]D 20 = -1 1.1 °, 589 nm, c = 0.37 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.98 min; MS (ESIpos): m/z = 551/553 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.55), 0.008 (1.55), 1.031 (0.68), 1.046 (0.70), 1.234 (0.66), 1.284 (1.07), 1.804 (0.44), 1.823 (0.94), 1.846 (1.23), 1.869 (0.68), 2.033 (0.59), 2.053 (2.43), 2.088 (2.71 ), 2.099 (2.69), 2.140 (4.66), 2.170 (1.18), 2.366 (0.81 ), 2.710 (0.85), 3.536 (0.42), 3.619 (1.01 ), 3.719 (1.42), 7.248 (0.88), 7.267 (1.86), 7.286 (1.34), 7.358 (1.16), 7.375 (2.06), 7.394 (1.09), 7.437 (3.50), 7.440 (3.33), 7.457 (2.89), 7.499 (6.46), 7.507 (4.44), 7.519 (16.00), 7.530 (5.52), 7.693 (0.46), 7.829 (2.23), 7.834 (1.99), 7.851 (3.31 ), 7.856 (3.20), 7.927 (6.22), 7.949 (3.92), 8.839 (1.23), 8.854 (2.39), 8.868 (1.16), 12.043 (1.79).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.05 (br. s, 1 H), 8.85 (t, 1 H), 7.94 (d, 1 H), 7.84 (dd, 1 H), 7.69 (br. s, 1 H), 7.56-7.47 (m, 6H), 7.45 (dd, 1 H), 7.38 (br. t, 1 H), 7.27 (br. t, 1 H), 3.83-3.53 (m, 3H), 2.22-2.00 (m, 6H), 1.92-1.74 (m, 1 H).
Beispiel 153
(+)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlorphenyl)pentansäure (Enantiomer 2)
Bei der in Beispiel 151 beschriebenen Enantiomerentrennung wurden 496 mg (98% Reinheit, ee-Wert 97%) der Titelverbindung als später eluierendes Enantiomer erhalten. Die Substanz wurde anschließend lyophilisiert. [a]D 20 = +16.5°, 589 nm, c = 0.44 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.98 min; MS (ESIpos): m/z = 551/553 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.95), 0.008 (0.81 ), 1.234 (0.60), 1.285 (0.86), 1.805 (0.48), 1.824 (1.03), 1.847 (1.34), 1.870 (0.74), 2.034 (0.66), 2.044 (0.81 ), 2.054 (2.62), 2.074 (2.15), 2.089 (2.92), 2.100 (2.92), 2.141 (5.04), 2.171 (1.25), 2.523 (0.48), 3.621 (1.09), 3.687 (0.58), 3.707 (1.13), 3.721 (1.56), 3.732 (1.34), 7.248 (0.92), 7.267 (1.98), 7.285 (1.39), 7.357 (1.22), 7.376 (2.16), 7.394 (1.15), 7.437 (3.50), 7.440 (3.25), 7.457 (2.93), 7.460 (2.67), 7.477 (0.67), 7.489 (1.92), 7.499 (6.75), 7.507 (4.65), 7.519 (16.00), 7.530 (5.84), 7.692 (0.46),
7.828 (2.25), 7.834 (1.98), 7.851 (3.36), 7.856 (3.13), 7.927 (6.00), 7.949 (3.83), 8.840 (1.33), 8.855 (2.61 ), 8.869 (1.26), 12.045 (1.75).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.05 (br. s, 1 H), 8.85 (t, 1 H), 7.94 (d, 1 H), 7.84 (dd, 1 H), 7.69 (br. s, 1 H), 7.55-7.48 (m, 6H), 7.45 (dd, 1 H), 7.38 (br. t, 1 H), 7.27 (br. t, 1 H), 3.83-3.55 (m, 3H), 2.22-2.01 (m, 6H), 1.91-1.77 (m, 1 H).
Beispiel 154
(+/-)-5-{[(6-Brom-3-methyl-2-phenylcN^
pentansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 4-(2-chlorphenyl)-4-methylpentanoat (80 mg, 129 μηηοΙ, Beispiel 233A) in Dichlormethan (91 1 μΙ) wurde TFA (198 μΙ, 2.57 mmol) gegeben, und das Gemisch wurde 7 h bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 68 mg (98% Reinheit, 92% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.07 min; MS (ESIpos): m/z = 565/567 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (1.35), 1.382 (0.70), 1.521 (16.00), 1.725 (0.83), 1.737 (0.96), 1.754 (1.30), 1.765 (1.99), 1.776 (1.15), 1.793 (1.58), 1.805 (1.47), 1.889 (1.36), 1.901 (1.55), 1.923 (1.80), 1.934 (1.86), 1.952 (0.95), 1.964 (1.39), 2.006 (1.79), 2.018 (1.52), 2.036 (2.12), 2.046 (2.66), 2.056 (1.85), 2.074 (2.33), 2.086 (2.02), 2.312 (0.43), 2.327 (0.54), 2.366 (0.43), 2.523 (1.26), 2.648 (0.89), 2.660 (1.12), 2.679 (1.55), 2.690 (1.43), 2.710 (1.15), 2.722 (0.71 ), 3.670 (1.74), 3.683 (1.89), 3.704 (2.07), 3.717 (1.88), 4.342 (2.29), 4.359 (2.49), 4.375 (2.34), 4.393 (2.19), 7.261 (0.91 ), 7.278 (2.37), 7.302 (2.68), 7.325 (3.06), 7.341 (1.52), 7.414 (4.36), 7.417 (4.44), 7.432 (4.91 ), 7.446 (2.34), 7.473 (0.77), 7.489 (1.89), 7.499 (6.18), 7.506 (5.65), 7.518 (14.98), 7.522 (13.05), 7.665 (0.52), 7.828 (2.74), 7.833 (2.49), 7.850 (4.25), 7.855 (4.03), 7.924 (7.73), 7.946 (4.92), 8.623 (1.86), 8.638 (3.12), 8.653 (1.90).
Trennunq der Enantiomere:
Die Titelverbindung (55 mg) wurde in Methanol (10 ml) gelöst und mittels praparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 155 und 156) [Säule: Daicel Chiral- pak AD SFC, 5 μηι, 250 mm x 20 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 0.5 ml; Eluent: 80% Kohlendioxid / 20% Ethanol; Laufzeit 18 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet.
Beispiel 155
(-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlorphenyl)-4-methyl- pentansäure (Enantiomer 1)
Bei der in Beispiel 154 beschriebenen Enantiomerentrennung wurden 17 mg (98% Reinheit, ee-Wert 100%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -38.2°, 589 nm, c = 0.27 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.01 min; MS (ESIpos): m/z = 565/567 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.234 (1.04), 1.520 (16.00), 1.721 (0.73), 1.732 (0.85), 1.750 (1.20), 1.760 (1.83), 1.770 (1.10), 1.788 (1.39), 1.800 (1.29), 1.889 (1.23), 1.901 (1.42), 1.923 (1.74), 1.933 (1.83), 1.952 (0.95), 1.964 (1.36), 2.001 (1.61 ), 2.013 (1.39), 2.031 (1.96), 2.040 (2.43), 2.069 (1.99), 2.080 (1.89), 2.310 (0.50), 2.328 (0.54), 2.366 (1.55), 2.643 (0.85), 2.655 (1.01 ), 2.675 (1.74), 2.709 (2.15), 3.313 (12.37), 3.670 (1.67), 3.682 (1.83), 3.703 (1.96), 3.716 (1.80), 4.341 (1.89), 4.358 (2.08), 4.374 (1.89), 4.392 (1.74), 7.259 (1.01 ), 7.277 (2.52), 7.301 (2.81 ), 7.324 (3.22), 7.340 (1.48), 7.417 (4.42), 7.432 (4.70), 7.446 (2.56), 7.473 (0.88), 7.488 (2.02), 7.498 (6.28), 7.505 (5.78), 7.516 (14.93), 7.521 (13.32), 7.654 (0.54), 7.825 (2.65), 7.830 (2.52), 7.848 (4.07), 7.853 (4.04), 7.922 (7.57), 7.945 (4.83), 8.631 (1.77), 8.646 (3.00), 8.661 (1.70).
Beispiel 156
(+)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlorphenyl)-4-methyl- pentansäure (Enantiomer 2)
Bei der in Beispiel 154 beschriebenen Enantiomerentrennung wurden 17 mg (98% Reinheit, ee-Wert 100%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = +40.0°, 589 nm, c = 0.27 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.01 min; MS (ESIpos): m/z = 565/567 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.235 (0.76), 1.520 (16.00), 1.719 (0.74), 1.730 (0.82), 1.758 (1.74), 1.786 (1.33), 1.797 (1.22), 1.889 (1.19), 1.900 (1.38), 1.931 (1.86), 1.963 (1.32), 1.999 (1.58), 2.01 1 (1.36), 2.039 (2.39), 2.067 (1.93), 2.078 (1.94), 2.309 (0.52), 2.327 (0.73),
2.653 (1.00), 2.672 (1.91 ), 2.709 (0.93), 3.669 (1.69), 3.682 (1.82), 3.702 (1.98), 3.715 (1.82), 4.341 (1.87), 4.357 (2.07), 4.374 (1.90), 4.391 (1.75), 7.260 (1.10), 7.277 (2.64), 7.300 (2.91 ), 7.324 (3.28), 7.340 (1.53), 7.417 (4.54), 7.432 (4.81 ), 7.472 (0.95), 7.497 (6.67), 7.505 (6.14), 7.516 (15.64), 7.670 (0.59), 7.825 (2.88), 7.830 (2.70), 7.847 (4.40), 7.852 (4.24), 7.922 (8.20), 7.945 (5.18), 8.635 (1.78), 8.649 (3.02), 8.664 (1.67).
Beispiel 157
(+/-)-5-{[(6-Brom-3-methyl-2-phenyl^
säure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 4-(2-chlor-6-fluorphenyl)pentanoat (550 mg, 879 μηηοΙ, Beispiel 236A) in Dichlormethan (8.3 ml) wurde TFA (1.4 ml, 18 mmol) gegeben, und das Gemisch wurde 16 h bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 499 mg (98% Reinheit, 98% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.04 min; MS (ESIpos): m/z = 569/571 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (1.37), 1.965 (0.62), 2.073 (2.03), 2.085 (2.50), 2.1 10 (2.88), 2.121 (2.94), 2.159 (8.08), 3.749 (1.78), 4.366 (0.42), 7.191 (0.74), 7.205 (1.1 1 ), 7.215 (1.29), 7.227 (1.07), 7.242 (1.01 ), 7.332 (4.41 ), 7.480 (0.49), 7.492 (1.47), 7.503 (4.44), 7.51 1 (4.26), 7.522 (16.00), 7.533 (6.25), 7.838 (2.44), 7.844 (2.21 ), 7.861 (3.71 ), 7.866 (3.56), 7.933 (6.68), 7.955 (4.19), 8.928 (1.23), 8.942 (2.23).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.94 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.72 (br. s, 1 H), 7.56-7.47 (m, 5H), 7.38-7.28 (m, 2H), 7.26-7.16 (m, 1 H), 3.91-3.68 (m, 3H), 2.25-1.88 (m, 7H).
Trennunq der Enantiomere:
Die Titelverbindung (440 mg) wurde in 40 ml eines 1 :1 -Gemisches aus Methanol und Acetonitril gelöst und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 158 und 159) [Säule: Daicel Chiralpak AD-H, 5 μηι, 250 mm x 30 mm; Fluss: 100 ml/min; Detektion: 210 nm; Temperatur: 38 °C; Injektion: 1.2 ml; Eluent: 68% Kohlendioxid / 32% Isopro- panol; Laufzeit 16 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet.
Beispiel 158
(-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlor-6-fluorphenyl)pentan- säure (Enantiomer 1)
Bei der in Beispiel 157 beschriebenen Enantiomerentrennung wurden 221 mg (98% Reinheit, ee-Wert 94%) der Titelverbindung als früher eluierendes Enantiomer erhalten. Die Substanz wurde anschließend lyophilisiert.
[a]D 20 = -28.1 °, 589 nm, c = 0.35 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.04 min; MS (ESIpos): m/z = 569/571 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.87), 0.008 (1.61 ), 1.030 (0.60), 1.046 (0.64), 1.966 (0.58), 2.084 (2.34), 2.109 (2.76), 2.1 19 (2.78), 2.158 (7.85), 2.524 (1.21 ), 3.748 (1.70), 7.190 (0.73), 7.204 (1.10), 7.214 (1.26), 7.227 (1.07), 7.242 (1.02), 7.331 (4.34), 7.478 (0.51 ), 7.491 (1.46), 7.501 (4.44), 7.509 (4.32), 7.521 (16.00), 7.527 (7.90), 7.532 (6.22), 7.535 (5.69), 7.836 (2.58), 7.841 (2.27), 7.858 (3.86), 7.864 (3.65), 7.931 (6.90), 7.953 (4.31 ), 8.926 (1.23), 8.940 (2.16), 12.075 (0.53).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.08 (br. s, 1 H), 8.94 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.72 (br. s, 1 H), 7.56-7.46 (m, 5H), 7.37-7.28 (m, 2H), 7.27-7.17 (m, 1 H), 3.94-3.66 (m, 3H), 2.23-1.89 (m, 7H). Beispiel 159
(+)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlor-6-fluorphenyl)pentan- säure (Enantiomer 2)
Bei der in Beispiel 157 beschriebenen Enantiomerentrennung wurden 192 mg (98% Reinheit, ee-Wert 94%) der Titelverbindung als später eluierendes Enantiomer erhalten. Die Substanz wurde anschließend lyophilisiert.
[a]D 20 = +38.2°, 589 nm, c = 0.36 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.04 min; MS (ESIpos): m/z = 569/571 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (1.41 ), 1.030 (0.64), 1.045 (0.68), 1.235 (0.53), 1.974 (0.64), 2.083 (2.36), 2.108 (2.73), 2.120 (2.76), 2.157 (7.76), 2.328 (0.54), 2.670 (0.42), 3.749 (1.71 ), 7.214 (1.20), 7.227 (1.08), 7.242 (1.01 ), 7.331 (4.31 ), 7.491 (1.41 ), 7.501 (4.31 ), 7.509 (4.22), 7.521 (16.00), 7.532 (6.07), 7.836 (2.45), 7.841 (2.22), 7.858 (3.75), 7.863 (3.52), 7.930 (6.91 ), 7.953 (4.26), 8.940 (2.16), 12.061 (0.45).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.06 (br. s, 1 H), 8.94 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.70 (br. s, 1 H), 7.56-7.46 (m, 5H), 7.38-7.29 (m, 2H), 7.27-7.17 (m, 1 H), 3.93-3.66 (m, 3H), 2.22-1.91 (m, 7H).
Beispiel 160
(+/-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-methyl^
(Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 4-(2-methylphenyl)pentanoat (75 mg, 98% Reinheit, 125 μηηοΙ, Beispiel 237A) in Dichlormethan (890 μΙ) wurde TFA (190 μΙ, 2.5 mmol) gegeben, und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 53 mg (98% Reinheit, 77% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.00 min; MS (ESIpos): m/z = 531/533 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.21 ), 0.008 (1.47), 1.777 (0.46), 1.793 (0.92), 1.818 (1.16), 1.837 (0.65), 2.026 (0.86), 2.044 (1.56), 2.053 (1.38), 2.073 (7.16), 2.082 (4.15), 2.107 (3.35), 2.331 (16.00), 2.523 (0.68), 3.320 (1.12), 3.338 (1.22), 3.367 (0.50), 3.478 (1.02),
3.490 (1.29), 3.510 (1.62), 3.524 (1.69), 3.540 (1.39), 3.714 (2.22), 3.733 (2.42), 3.749 (2.03), 3.765 (1.75), 3.784 (1.14), 3.864 (0.50), 7.095 (0.75), 7.1 13 (2.01 ), 7.131 (1.76), 7.167 (3.35),
7.184 (1.90), 7.194 (1.17), 7.214 (1.95), 7.232 (1.07), 7.304 (3.1 1 ), 7.323 (2.15), 7.477 (0.44),
7.491 (1.29), 7.501 (4.06), 7.509 (3.74), 7.521 (12.71 ), 7.533 (5.68), 7.685 (0.58), 7.834 (2.00),
7.839 (1.81 ), 7.857 (3.08), 7.862 (2.94), 7.933 (5.59), 7.955 (3.59), 8.827 (1.27), 8.842 (1.97), 8.856 (1.25).
Beispiel 161
(■)-5-{[(6-Brom-3-methyl-2-phenylch^
(Enantiomer 1)
Zu einer Lösung aus (-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4- (2-methylphenyl)pentanoat (1 10 mg, 187 μηηοΙ, Beispiel 238A) in Dichlormethan (1.3 ml) wurde TFA (290 μΙ, 3.7 mmol) gegeben, und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder ein- geengt. Der Rückstand wurde mittels präparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 85 mg (98% Reinheit, ee-Wert 99%, 84% d. Th.) der Titelverbindung erhalten.
[a]D 20 = -14.4°, 589 nm, c = 0.36 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.05 min; MS (ESIpos): m/z = 531/533 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (0.72), 1.762 (0.40), 1.778 (0.57), 1.793 (1.03), 1.817 (1.22), 1.837 (0.68), 2.026 (1.10), 2.045 (1.96), 2.054 (1.92), 2.073 (7.90), 2.082 (4.58), 2.100 (3.49), 2.107 (3.68), 2.283 (0.50), 2.299 (0.90), 2.331 (16.00), 2.523 (1.16), 3.320 (1.18), 3.338 (1.22), 3.367 (0.40), 3.478 (0.84), 3.490 (1.08), 3.510 (1.29), 3.524 (1.25), 3.539 (0.76), 3.715 (0.95), 3.733 (1.43), 3.750 (1.25), 3.766 (1.1 1 ), 3.785 (0.63), 7.095 (0.90), 7.1 13 (2.15), 7.131 (1.88), 7.167 (3.49), 7.184 (2.07), 7.194 (1.30), 7.213 (2.03), 7.231 (1.10), 7.304 (3.20), 7.323 (2.18), 7.478 (0.88), 7.491 (2.00), 7.501 (4.88), 7.509 (4.69), 7.521 (13.16), 7.533 (5.93), 7.686 (0.65), 7.834 (2.17), 7.840 (1.93), 7.857 (3.18), 7.862 (2.93), 7.933 (5.55), 7.955 (3.53), 8.827 (1.43), 8.841 (2.07), 8.855 (1.26).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1 1.98 (br. s, 1 H), 8.84 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.69 (br. s, 1 H), 7.56-7.45 (m, 5H), 7.34-7.28 (m, 1 H), 7.26-7.06 (m, 3H), 3.81-3.69 (m, 1 H), 3.56-3.46 (m, 1 H), 3.39-3.26 (m, 1 H), 2.33 (s, 3H), 2.18-1.98 (m, 6H), 1.87-1.74 (m, 1 H).
Beispiel 162
(+)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-methylphenyl)pentansäure (Enantiomer 2)
Zu einer Lösung aus (+)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4- (2-methylphenyl)pentanoat (1 10 mg, 187 μηηοΙ, Beispiel 239A) in Dichlormethan (1.3 ml) wurde TFA (290 μΙ, 3.7 mmol) gegeben, und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde erneut TFA (140 μΙ, 1.9 mmol) hinzugegeben, und das Gemisch wurde eine weitere Nacht bei RT gerührt. Danach wurde das Gemisch eingeengt und mehrmals mit Dichlormethan
versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 88 mg (98% Reinheit, ee-Wert 99%, 86% d. Th. der Titelverbindung erhalten.
[a]D 20 = +15.5°, 589 nm, c = 0.37 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.05 min; MS (ESIpos): m/z = 531/533 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (0.97), 1.778 (0.45), 1.793 (0.92), 1.818 (1.14), 1.837 (0.65), 2.026 (0.83), 2.044 (1.53), 2.053 (1.37), 2.073 (7.18), 2.082 (4.17), 2.107 (3.40), 2.331 (16.00), 2.523 (0.69), 3.320 (1.07), 3.338 (1.15), 3.366 (0.40), 3.478 (0.74), 3.490 (1.00), 3.510 (1.23), 3.524 (1.22), 3.539 (0.77), 3.714 (0.92), 3.733 (1.43), 3.750 (1.28), 3.766 (1.19), 3.785 (0.75), 7.095 (0.76), 7.113 (2.03), 7.131 (1.77), 7.167 (3.35), 7.184 (1.93), 7.194 (1.17), 7.214 (1.92), 7.231 (1.05), 7.304 (3.1 1 ), 7.323 (2.14), 7.478 (0.50), 7.488 (1.24), 7.491 (1.42), 7.501 (4.18), 7.509 (3.98), 7.521 (12.74), 7.532 (5.63), 7.535 (4.82), 7.543 (1.04), 7.685 (0.58), 7.834 (2.12), 7.839 (1.87), 7.857 (3.22), 7.862 (3.01 ), 7.933 (5.66), 7.955 (3.64), 8.827 (1.30), 8.841 (1.98), 8.855 (1.22).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.01 (br. s, 1 H), 8.84 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.69 (br. s, 1 H), 7.55-7.47 (m, 5H), 7.35-7.28 (m, 1 H), 7.25-7.08 (m, 3H), 3.80-3.69 (m, 1 H), 3.56-3.45 (m, 1 H), 3.39-3.27 (m, 1 H), 2.33 (s, 3H), 2.19-2.00 (m, 6H), 1.86-1.74 (m, 1 H).
Beispiel 163
(+/-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlor-5-fluorphenyl)pentan- säure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 4-(2-chlor-5-fluorphenyl)pentanoat (60 mg, 95.9 μηηοΙ, Beispiel 240A) in Dichlormethan (680 μΙ) wurde TFA (150 μΙ, 1.9 mmol) gegeben, und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde erneut TFA (150 μΙ, 1.9 mmol) hinzugegeben, und das Gemisch wurde weitere 24 h bei RT gerührt. Danach wurde das Gemisch eingeengt und mehrmals mit Dichlorme-
than versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 48 mg (98% Reinheit, 86% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.00 min; MS (ESIpos): m/z = 569/571 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.819 (1.01 ), 1.839 (1.23), 2.031 (0.98), 2.062 (2.52), 2.088 (2.22), 2.101 (2.02), 2.110 (2.52), 2.140 (8.20), 2.158 (2.61 ), 2.181 (0.91 ), 3.612 (1.09), 3.739 (2.47), 4.055 (1.13), 7.119 (0.79), 7.139 (1.42), 7.409 (1.76), 7.416 (1.87), 7.434 (1.85), 7.442 (1.75), 7.481 (2.77), 7.494 (3.57), 7.502 (5.96), 7.509 (4.48), 7.520 (16.00), 7.531 (5.82), 7.663 (0.43), 7.833 (2.34), 7.838 (2.1 1 ), 7.855 (3.52), 7.860 (3.44), 7.930 (6.44), 7.952 (4.06), 8.847 (1.27), 8.862 (2.54), 8.876 (1.24).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.86 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.66 (br. s, 1 H), 7.56-7.47 (m, 6H), 7.43 (dd, 1 H), 7.18-7.09 (m, 1 H), 3.79-3.55 (m, 3H), 2.22-1.99 (m, 6H), 1.88- 1.76 (m, 1 H).
Beispiel 164
(-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlor-5-fluorphenyl)pentan- säure (Enantiomer 1)
Zu einer Lösung aus (-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4- (2-chlor-5-fluorphenyl)pentanoat (1.56 g, 86% Reinheit, 2.14 mmol, Beispiel 241 A) in Dichlormethan (15 ml) wurde TFA (3.3 ml, 43 mmol) gegeben, und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde zweimal mittels praparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 949 mg (98% Reinheit, ee-Wert 99%, 76% d. Th.) der Titelverbindung erhalten. [a]D 20 = -22.2°, 589 nm, c = 0.42 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.97 min; MS (ESIpos): m/z = 569/571 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.07), 1.797 (0.50), 1.819 (1.05), 1.840 (1.25), 1.861 (0.74), 2.018 (0.76), 2.031 (1.01 ), 2.050 (1.19), 2.061 (2.61 ), 2.073 (0.72), 2.088 (2.28), 2.101 (2.05), 2.1 10 (2.52), 2.140 (8.45), 2.158 (2.74), 2.181 (0.93), 2.198 (0.44), 3.615 (1.02), 3.738 (2.40), 7.1 19 (0.79), 7.139 (1.41 ), 7.154 (0.82), 7.408 (1.88), 7.416 (1.91 ), 7.434 (1.91 ), 7.441 (1.83), 7.480 (2.72), 7.494 (3.60), 7.502 (5.20), 7.508 (4.41 ), 7.519 (16.00), 7.529 (5.73), 7.830 (2.18), 7.835 (2.03), 7.853 (3.29), 7.858 (3.25), 7.928 (5.99), 7.950 (3.83), 8.845 (1.32), 8.860 (2.66), 8.874 (1.26), 12.066 (1.15).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.07 (br. s, 1 H), 8.86 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.66 (br. s, 1 H), 7.55-7.47 (m, 6H), 7.42 (dd, 1 H), 7.18-7.09 (m, 1 H), 3.82-3.55 (m, 3H), 2.21-1.98 (m, 6H), 1.91 -1.76 (m, 1 H).
Beispiel 165
(+)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-ch
säure (Enantiomer 2)
Zu einer Lösung aus (+)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4- (2-chlor-5-fluorphenyl)pentanoat (1.26 g, 98% Reinheit, 1.97 mmol, Beispiel 242A) in Dichlor- methan (14 ml) wurde TFA (3.0 ml, 39 mmol) gegeben, und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 1.03 g (98% Reinheit, ee-Wert 95%, 89% d. Th.) der Titelverbindung erhalten.
[a]D 20 = +20.1 °, 589 nm, c = 0.38 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.97 min; MS (ESIpos): m/z = 569/571 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.01 ), 0.008 (1.76), 1.796 (0.49), 1.819 (1.03), 1.828 (0.64), 1.840 (1.24), 1.861 (0.71 ), 2.018 (0.76), 2.031 (1.00), 2.049 (1.19), 2.061 (2.63), 2.073 (0.82), 2.088 (2.31 ), 2.101 (2.07), 2.1 10 (2.51 ), 2.140 (8.28), 2.158 (2.73), 2.181 (0.94), 2.198 (0.45), 2.523 (0.64), 3.616 (1.00), 3.738 (2.38), 7.1 19 (0.77), 7.139 (1.37), 7.154 (0.82), 7.408 (1.85), 7.416 (1.90), 7.434 (1.90), 7.441 (1.82), 7.480 (2.76), 7.494 (3.67), 7.500 (5.17), 7.508 (4.41 ), 7.519 (16.00), 7.530 (5.70), 7.830 (2.31 ), 7.835 (2.07), 7.853 (3.49), 7.858 (3.30), 7.928 (6.23), 7.950 (3.98), 8.845 (1.30), 8.860 (2.64), 8.874 (1.26), 12.067 (1.03).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.07 (br. s, 1 H), 8.86 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.66 (br. s, 1 H), 7.56-7.47 (m, 6H), 7.42 (dd, 1 H), 7.18-7.10 (m, 1 H), 3.83-3.54 (m, 3H), 2.22-1.99 (m, 6H), 1.90-1.76 (m, 1 H).
Beispiel 166
(+/-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-methoxyphenyl)pentan- säure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 4-(2-methoxyphenyl)pentanoat (40 mg, 98% Reinheit, 64.9 μηηοΙ, Beispiel 243A) in Dichlormethan (460 μΙ) wurde TFA (100 μΙ, 1.3 mmol) gegeben, und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 35 mg (98% Reinheit, 96% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.96 min; MS (ESIpos): m/z = 547/549 [M+H]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.195 (0.72), 1.212 (0.73), 1.776 (0.74), 1.802 (1.20), 1.816 (1.27), 1.853 (0.50), 1.985 (1.24), 2.004 (1.49), 2.016 (2.88), 2.049 (4.81 ), 2.075 (3.56), 2.092 (1.80), 2.146 (7.88), 2.328 (0.67), 2.367 (0.76), 2.670 (0.44), 2.731 (1.81 ), 2.890 (2.22), 3.401 (1.49), 3.620 (0.86), 3.636 (1.30), 3.653 (1.88), 3.668 (2.28), 3.683 (1.42), 3.704 (1.55), 3.721 (2.03), 3.740 (2.03), 4.988 (0.72), 6.927 (1.69), 6.946 (3.49), 6.964 (2.23), 6.979 (3.61 ), 6.999 (4.03), 7.200 (1.99), 7.220 (3.32), 7.228 (4.01 ), 7.246 (3.29), 7.492 (2.1 1 ), 7.502 (5.02), 7.510 (5.09), 7.522 (16.00), 7.707 (1.77), 7.832 (2.38), 7.838 (2.30), 7.855 (3.59), 7.860 (3.61 ), 7.928 (6.05), 7.950 (4.12), 8.757 (1.62), 8.772 (2.96), 8.786 (1.60).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.77 (br. t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.71 (br. s, 1 H), 7.57-7.46 (m, 5H), 7.27-7.18 (m, 2H), 7.02-6.89 (m, 2H), 3.78 (s, 3H), 3.77-3.60 (m, 2H), 3.46-3.34 (m, 1 H), 2.19-1.94 (m, 6H), 1.88-1.73 (m, 1 H).
Beispiel 167
(-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-methoxyphenyl)pentansäure (Enantiomer 1)
Zu einer Lösung aus (-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4- (2-methoxyphenyl)pentanoat (60 mg, 98% Reinheit, 97.4 μηηοΙ, Beispiel 244A) in Dichlormethan (690 μΙ) wurde TFA (150 μΙ, 1.9 mmol) gegeben, und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde erneut TFA (150 μΙ, 1.9 mmol) hinzugegeben, und das Gemisch wur-
de weitere 3 h bei RT gerührt. Danach wurde das Gemisch eingeengt und mehrmals mit Dich- lormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert. Es wurden 51 mg (98% Reinheit, ee-Wert 97%, 93% d. Th.) der Titelverbindung erhalten. [a]D 20 = -12.6°, 589 nm, c = 0.37 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.05 min; MS (ESIpos): m/z = 547/549 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.92), 0.008 (1.56), 1.776 (0.63), 1.801 (1.03), 1.815 (1.14), 1.829 (0.88), 1.839 (0.64), 1.853 (0.42), 1.984 (0.94), 1.997 (1.05), 2.004 (1.10), 2.016 (2.66), 2.043 (3.49), 2.048 (4.43), 2.074 (3.42), 2.092 (1.49), 2.1 14 (0.98), 2.145 (7.48), 2.523 (1.02), 3.399 (1.46), 3.599 (0.77), 3.620 (1.26), 3.634 (1.77), 3.652 (2.54), 3.667 (3.10), 3.682 (2.34), 3.703 (2.65), 3.719 (3.18), 3.739 (3.19), 3.756 (2.77), 3.953 (0.59), 6.927 (1.56), 6.945 (3.49), 6.964 (2.06), 6.979 (3.60), 6.998 (4.19), 7.200 (1.81 ), 7.219 (3.06), 7.227 (3.89), 7.239 (1.91 ), 7.246 (3.28), 7.477 (0.49), 7.490 (1.47), 7.500 (4.51 ), 7.508 (4.29), 7.520 (16.00), 7.531 (6.55), 7.705 (1.50), 7.829 (2.44), 7.834 (2.20), 7.851 (3.77), 7.856 (3.62), 7.926 (6.85), 7.948 (4.37), 8.755 (1.48), 8.770 (2.95), 8.784 (1.47).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.00 (br. s, 1 H), 8.77 (t, 1 H), 7.93 (d, 1 H), 7.85 (dd, 1 H), 7.71 (br. s, 1 H), 7.55-7.46 (m, 5H), 7.27-7.18 (m, 2H), 7.01-6.91 (m, 2H), 3.78 (s, 3H), 3.77- 3.61 (m, 2H), 3.46-3.33 (m, 1 H), 2.19-1.95 (m, 6H), 1.87-1.74 (m, 1 H).
Beispiel 168
(+)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-methoxyphenyl)pentan- säure (Enantiomer 2)
Zu einer Lösung aus (+)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4- (2-methoxyphenyl)pentanoat (55 mg, 98% Reinheit, 89.3 μηηοΙ, Beispiel 245A) in Dichlormethan (630 μΙ) wurde TFA (140 μΙ, 1.8 mmol) gegeben, und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde erneut TFA (140 μΙ, 1.8 mmol) hinzugegeben, und das Gemisch wurde weitere 4 h bei RT gerührt. Danach wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert. Es wurden 46 mg (98% Reinheit, ee-Wert 99%, 92% d. Th.) der Titelverbindung erhal- ten.
[a]D 20 = +14.1 °, 589 nm, c = 0.35 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.05 min; MS (ESIpos): m/z = 547/549 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.42), 1.802 (0.63), 1.815 (0.70), 1.828 (0.53), 1.984 (0.57), 1.997 (0.64), 2.004 (0.65), 2.016 (1.63), 2.043 (2.14), 2.048 (2.70), 2.074 (2.10),
2.092 (0.92), 2.1 14 (0.59), 2.145 (4.55), 2.524 (0.53), 3.400 (1.05), 3.431 (0.60), 3.619 (2.56), 3.633 (2.25), 3.652 (2.13), 3.667 (2.07), 3.682 (1.32), 3.703 (1.21 ), 3.719 (1.35), 3.739 (1.22), 3.756 (0.91 ), 3.776 (16.00), 6.927 (0.96), 6.946 (2.13), 6.964 (1.26), 6.979 (2.22), 6.998 (2.60), 7.200 (1.12), 7.219 (1.89), 7.227 (2.40), 7.239 (1.18), 7.246 (2.03), 7.490 (0.90), 7.500 (2.81 ), 7.508 (2.70), 7.520 (9.88), 7.527 (5.03), 7.531 (4.00), 7.534 (3.68), 7.704 (0.90), 7.829 (1.57), 7.834 (1.40), 7.851 (2.41 ), 7.856 (2.30), 7.926 (4.33), 7.948 (2.78), 8.755 (0.91 ), 8.770 (1.80), 8.784 (0.90).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1 1.97 (br. s, 1 H), 8.77 (t, 1 H), 7.93 (d, 1 H), 7.85 (dd, 1 H), 7.70 (br. s, 1 H), 7.56-7.44 (m, 5H), 7.26-7.18 (m, 2H), 7.02-6.91 (m, 2H), 3.78 (s, 3H), 3.76- 3.61 (m, 2H), 3.45-3.35 (m, 1 H), 2.20-1.94 (m, 6H), 1.87-1.73 (m, 1 H).
Beispiel 169
(+/-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-^
methylpentansäure (Racemat)

Zu einer Lösung aus (+/-)-ierf-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 4-(2-chlor-5-fluorphenyl)-4-methylpentanoat (40 mg, 72% Reinheit, 45.0 μηηοΙ, Beispiel 246A) in Dichlormethan (320 μΙ) wurde TFA (69 μΙ, 900 μηηοΙ) gegeben, und das Gemisch wurde 48 h bei RT gerührt. Anschließend wurde erneut TFA (69 μΙ, 900 μηηοΙ) hinzugegeben, und das Gemisch wurde weitere 24 h bei RT gerührt. Danach wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 15) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand mit Ethylacetat und gesättigter Natriumhydrogencarbonat-Lösung (jeweils 10 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase zweimal mit Ethylacetat (jeweils 10 ml) extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, fil- triert und eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 25 mg (98% Reinheit, 93% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): R
t = 1.09 min; MS (ESIpos): m/z = 583/585 [M+H]
+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.233 (0.75), 1.334 (0.50), 1.475 (16.00), 1.555 (0.62), 1.592 (1.31 ), 1.617 (0.98), 1.853 (0.95), 1.877 (1.37), 1.914 (1.56), 1.951 (1.24), 2.146 (2.24), 2.259 (0.40), 2.327 (0.62), 2.366 (0.44), 2.669 (0.59), 2.710 (0.42), 3.327 (4.51 ), 3.698 (1.25), 4.301 (1.03), 7.1 19 (1.15), 7.139 (2.08), 7.160 (1.25), 7.217 (2.04), 7.246 (2.10), 7.445 (1.49), 7.468 (1.99), 7.484 (2.99), 7.499 (6.68), 7.504 (5.17), 7.518 (9.57), 7.532 (1 1.52), 7.644 (0.86),
7.820 (2.62), 7.825 (2.41 ), 7.842 (3.95), 7.847 (3.88), 7.921 (7.1 1 ), 7.943 (4.62), 9.1 10 (0.51 ).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 9.1 1 (br. s, 1 H), 7.93 (d, 1 H), 7.84 (dd, 1 H), 7.64 (br. s, 1 H), 7.57-7.42 (m, 6H), 7.23 (br. d, 1 H), 7.18-7.07 (m, 1 H), 4.39-4.25 (m, 1 H), 3.77-3.62 (m, 1 H), 2.49-2.40 (m, 1 H, teilweise verdeckt), 2.15 (br. s, 3H), 2.02-1.79 (m, 2H), 1.67-1.52 (m, 1 H), 1.47 (s, 3H).
Beispiel 170
(■)_5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlor-5-fluorpheny methylpentansäure (Enantiomer 1)
Zu einer Lösung aus (-)-ierf-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2- chlor-5-fluorphenyl)-4-methylpentanoat (60 mg, 93.8 μηηοΙ, Beispiel 247A) in Dichlormethan (690 μΙ) wurde TFA (72 μΙ, 940 μηηοΙ) gegeben, und das Gemisch wurde über Nacht bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand mit Ethylacetat und gesättigter Natriumhydrogencarbonat-Lösung (jeweils 10 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase zweimal mit Ethylacetat (jeweils 10 ml) extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 48 mg (98% Reinheit, 86% d. Th.) der Titelverbindung erhalten. [a]D 20 = -10.4°, 589 nm, c = 0.25 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.05 min; MS (ESIpos): m/z = 583/585 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.463 (16.00), 1.744 (1.05), 1.761 (1.23), 1.776 (0.90), 1.998 (1.25), 2.016 (1.10), 2.165 (5.53), 2.332 (1.41 ), 2.353 (1.27), 2.366 (1.37), 2.670 (0.74), 2.710 (0.64), 3.736 (0.94), 4.244 (0.86), 7.101 (1.05), 7.108 (1.30), 7.127 (2.15), 7.141 (1.37), 7.149 (1.42), 7.256 (1.82), 7.281 (1.92), 7.435 (1.75), 7.450 (2.12), 7.457 (2.03), 7.471 (2.15), 7.485 (2.13), 7.500 (7.01 ), 7.518 (8.09), 7.538 (9.99), 7.556 (2.99), 7.655 (3.04), 7.816 (2.46),
7.821 (2.31 ), 7.839 (3.72), 7.844 (3.66), 7.919 (6.26), 7.942 (4.16).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 9.45 (br. s, 1 H), 7.93 (d, 1 H), 7.83 (dd, 1 H), 7.65 (br. s, 1 H), 7.58-7.41 (m, 6H), 7.27 (br. d, 1 H), 7.16-7.08 (m, 1 H), 4.24 (br. s, 1 H), 3.74 (br. s, 1 H),
2.42-2.29 (m, 1 H), 2.17 (br. s, 3H), 2.07-1.93 (m, 1 H), 1.83-1.68 (m, 1 H), 1.46 (s, 3H), 1.55-1.43 (m, 1 H).
Beispiel 171
(+)-5-{[(6-Brom-3-methyl-2-phenylcN^
methylpentansäure (Enantiomer 2)
Zu einer Lösung aus (+)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4- (2-chlor-5-fluorphenyl)-4-methylpentanoat (80 mg, 125 μιτιοΙ, Beispiel 248A) in Dichlormethan (920 μΙ) wurde TFA (96 μΙ, 1.3 mmol) gegeben, und das Gemisch wurde über Nacht bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand mit Ethylacetat und gesättigter Natriumhydrogencarbonat-Lösung (jeweils 10 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase zweimal mit Ethylacetat (jeweils 10 ml) extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und einge- engt, und der Rückstand wurde lyophilisiert. Es wurden 68 mg (98% Reinheit, 91 % d. Th.) der Titelverbindung erhalten.
[a]D 20 = +14.5°, 589 nm, c = 0.25 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.05 min; MS (ESIpos): m/z = 583/585 [M+H]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.149 (0.46), 0.147 (0.46), 1.462 (16.00), 1.743 (1.16), 1.761 (1.28), 2.007 (1.21 ), 2.168 (6.38), 2.327 (1.98), 2.366 (1.39), 2.669 (1.02), 2.689 (2.41 ), 2.710 (0.78), 2.731 (0.91 ), 2.890 (0.88), 3.731 (0.89), 4.213 (0.83), 7.109 (1.31 ), 7.128 (2.20), 7.142 (1.34), 7.148 (1.37), 7.261 (1.83), 7.286 (1.85), 7.435 (1.90), 7.450 (2.20), 7.457 (2.12), 7.472 (2.30), 7.485 (2.25), 7.500 (7.08), 7.518 (7.99), 7.539 (9.68), 7.558 (2.92), 7.656 (3.56), 7.816 (2.46), 7.821 (2.36), 7.838 (3.76), 7.843 (3.65), 7.919 (6.25), 7.941 (4.07), 9.524 (0.41 ). 1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 9.52 (br. s, 1 H), 7.93 (d, 1 H), 7.83 (dd, 1 H), 7.66 (br. s, 1 H), 7.57-7.42 (m, 6H), 7.27 (br. d, 1 H), 7.16-7.09 (m, 1 H), 4.22 (br. s, 1 H), 3.73 (br. s, 1 H), 2.40-2.27 (m, 1 H), 2.17 (br. s, 3H), 2.07-1.95 (m, 1 H), 1.83-1.70 (m, 1 H), 1.49 (m, 1 H), 1.46 (s, 3H).
Beispiel 172
(+/-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlor-3,6-difluorphenyl)- pentansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 4-(2-chlor-3,6-difluorphenyl)pentanoat (100 mg, 155 μηιοΙ, Beispiel 249A) in Dichlormethan (1.1 ml) wurde TFA (120 μΙ, 1.6 mmol) gegeben, und das Gemisch wurde 5 Tage bei RT stehen ge- lassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert. Es wurden 81 mg (96% Reinheit, 85% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.07 min; MS (ESIpos): m/z = 587/589 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.48), 0.008 (2.23), 1.366 (1.46), 1.374 (1.01 ), 1.989 (0.64), 2.077 (1.02), 2.090 (1.26), 2.096 (1.29), 2.1 1 1 (2.28), 2.153 (8.77), 2.187 (1.59), 2.209 (0.60), 2.225 (0.43), 2.524 (0.98), 3.761 (1.41 ), 3.801 (1.14), 7.270 (0.60), 7.281 (0.72), 7.294 (1.33), 7.304 (1.38), 7.318 (1.03), 7.329 (0.90), 7.406 (1.25), 7.415 (1.25), 7.482 (0.54), 7.495 (1.55), 7.505 (4.63), 7.513 (4.55), 7.524 (16.00), 7.535 (6.42), 7.538 (5.67), 7.844 (2.39), 7.849 (2.16), 7.866 (3.69), 7.871 (3.52), 7.938 (6.67), 7.960 (4.19), 8.942 (1.21 ), 8.956 (2.17).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.96 (t, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.72 (br. s, 1 H), 7.56-7.48 (m, 5H), 7.45-7.37 (m, 1 H), 7.35-7.25 (m, 1 H), 3.95-3.68 (m, 3H), 2.26-1.92 (m, 7H).
Beispiel 173
(-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlor-3,6-difluorphenyl)- pentansäure (Enantiomer 1)
Zu einer Lösung aus (-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4- (2-chlor-3,6-difluorphenyl)pentanoat (240 mg, 373 μηηοΙ, Beispiel 250A) in Dichlormethan (2.7 ml) wurde TFA (290 μΙ, 3.7 mmol) gegeben, und das Gemisch wurde 2 Tage bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan ver- setzt und wieder eingeengt. Der Rückstand wurde in Acetonitril aufgenommen und mittels präparativer HPLC (Methode 15) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der
Rückstand lyophilisiert. Es wurden 212 mg (100% Reinheit, ee-Wert >95%, 97% d. Th.) der Titelverbindung erhalten.
[a]D 20 = -39.2°, 589 nm, c = 0.34 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.99 min; MS (ESIpos): m/z = 587/589 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (1.93), 1.990 (0.67), 2.073 (3.12), 2.097 (1.31 ), 2.1 12 (2.29), 2.154 (9.13), 2.187 (1.62), 2.210 (0.58), 2.524 (0.76), 3.762 (1.50), 3.803 (1.22), 4.388 (0.53), 7.270 (0.59), 7.281 (0.72), 7.294 (1.36), 7.304 (1.41 ), 7.319 (1.05), 7.330 (0.90), 7.405 (1.27), 7.415 (1.22), 7.482 (0.54), 7.495 (1.55), 7.505 (4.66), 7.513 (4.50), 7.524 (16.00), 7.535 (6.51 ), 7.843 (2.34), 7.849 (2.1 1 ), 7.866 (3.63), 7.871 (3.44), 7.938 (6.46), 7.960 (4.05), 8.942 (1.26), 8.957 (2.25).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1 1.97 (br. s, 1 H), 8.96 (t, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.72 (br. s, 1 H), 7.57-7.47 (m, 5H), 7.45-7.37 (m, 1 H), 7.35-7.24 (m, 1 H), 3.94-3.67 (m, 3H), 2.25-1.90 (m, 7H).
Beispiel 174
(+)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlor-3,6-difluorphenyl)- pentansäure (Enantiomer 2)
Zu einer Lösung aus (+)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4- (2-chlor-3,6-difluorphenyl)pentanoat (250 mg, 388 μηηοΙ, Beispiel 251A) in Dichlormethan (2.9 ml) wurde TFA (300 μΙ, 3.9 mmol) gegeben, und das Gemisch wurde 2 Tage bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde in Acetonitril aufgenommen und mittels präpa- rativer HPLC (Methode 15) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert. Es wurden 217 mg (100% Reinheit, ee-Wert 98%, 95% d. Th.) der Titelverbindung erhalten. [a]D 20 = +37.4°, 589 nm, c = 0.36 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.99 min; MS (ESIpos): m/z = 587/589 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (3.25), 0.008 (2.24), 1.990 (0.66), 2.073 (2.48), 2.097 (1.28), 2.1 1 1 (2.23), 2.153 (8.83), 2.186 (1.54), 2.209 (0.55), 2.524 (1.02), 3.740 (2.63), 7.270 (0.59), 7.281 (0.71 ), 7.294 (1.32), 7.304 (1.37), 7.319 (1.03), 7.329 (0.88), 7.405 (1.24), 7.415 (1.20), 7.494 (1.50), 7.504 (4.49), 7.512 (4.34), 7.523 (16.00), 7.535 (6.16), 7.842 (2.29), 7.847 (2.09), 7.864 (3.52), 7.870 (3.33), 7.936 (6.38), 7.959 (3.98), 8.940 (1.22), 8.955 (2.16).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.10 (br. s, 1 H), 8.95 (t, 1 H), 7.94 (d, 1 H), 7.86 (dd, 1 H), 7.72 (br. s, 1 H), 7.56-7.47 (m, 5H), 7.46-7.36 (m, 1 H), 7.34-7.26 (m, 1 H), 3.83-3.62 (m, 3H), 2.24-1.90 (m, 7H).
Beispiel 175
(+/-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amin
(Racemat)

Zu einer Lösung aus (+/-)-ierf-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 4-(2-fluorphenyl)pentanoat (500 mg, 845 μηηοΙ, Beispiel 252A) in Dichlormethan (6.2 ml) wurde TFA (650 μΙ, 8.5 mmol) gegeben, und das Gemisch wurde über Nacht bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde in Acetonitril aufgenommen und mittels praparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert. Das Lyophilisat wurde mit Ethylacetat und gesättigter Natriumhydrogencarbo- nat-Lösung (jeweils 10 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase zweimal mit Ethylacetat (jeweils 10 ml) extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde lyophil i- siert. Es wurden 449 mg (98% Reinheit, 97% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.90 min; MS (ESIpos): m/z = 535/537 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.788 (0.55), 1.805 (0.74), 1.814 (0.87), 1.828 (0.96), 1.839 (0.68), 1.850 (0.52), 2.033 (0.88), 2.045 (1.01 ), 2.060 (2.26), 2.069 (1.20), 2.086 (3.57), 2.094 (4.79), 2.1 17 (4.38), 2.134 (2.79), 2.155 (1.10), 3.347 (0.85), 3.359 (0.89), 3.368 (0.87), 3.637 (0.46), 3.651 (0.80), 3.670 (1.01 ), 3.684 (1.39), 3.697 (0.75), 3.735 (0.60), 3.753 (0.87), 3.771 (0.75), 7.144 (1.24), 7.166 (1.93), 7.191 (2.44), 7.208 (2.20), 7.226 (1.54), 7.271 (0.78), 7.286 (1.30), 7.302 (1.25), 7.324 (0.53), 7.401 (1.30), 7.404 (1.30), 7.419 (2.25), 7.438 (1.13), 7.490 (1.36), 7.502 (3.61 ), 7.510 (4.01 ), 7.520 (16.00), 7.834 (1.78), 7.839 (1.57), 7.857 (2.72), 7.861 (2.48), 7.929 (5.01 ), 7.951 (3.1 1 ), 8.853 (1.26), 8.868 (2.06), 8.882 (1.09).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.87 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.68 (br. s, 1 H), 7.55-7.46 (m, 5H), 7.45-7.38 (m, 1 H), 7.36-7.25 (m, 1 H), 7.25-7.13 (m, 2H), 3.83-3.72 (m, 1 H), 3.71-3.62 (m, 1 H), 3.41-3.30 (m, 1 H), 2.20-1.99 (m, 6H), 1.88-1.75 (m, 1 H).
Trennunq der Enantiomere:
Die Titelverbindung (400 mg) wurde in Methanol (20 ml) gelöst und mittels praparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 176 und 177) [Säule: Daicel Chiral- pak AD, 5 μηη, 250 mm x 20 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injek- tion: 1.0 ml; Eluent: 21 % Ethanol / 79% Kohlendioxid; Laufzeit 13 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet.
Beispiel 176
(-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-fluorphenyl)pentansäure (Enantiomer 1)
Bei der in Beispiel 175 beschriebenen Enantiomerentrennung wurden 128 mg (98% Reinheit, ee-Wert 96%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -32.2°, 589 nm, c = 0.34 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.00 min; MS (ESIpos): m/z = 535/537 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.76), 0.008 (1.82), 1.785 (0.53), 1.812 (0.78), 1.826 (0.88), 1.847 (0.53), 2.030 (1.00), 2.056 (2.16), 2.073 (2.12), 2.083 (3.51 ), 2.092 (4.44), 2.1 14 (4.01 ), 2.131 (2.68), 2.523 (1.54), 3.530 (2.19), 3.634 (0.52), 3.648 (0.83), 3.667 (1.03), 3.681 (1.32), 3.695 (0.75), 3.732 (0.63), 3.750 (0.85), 7.144 (1.22), 7.165 (1.92), 7.189 (2.26), 7.206 (2.10), 7.225 (1.45), 7.285 (1.19), 7.302 (1.04), 7.399 (1.28), 7.418 (2.19), 7.436 (1.19), 7.486 (1.67), 7.499 (3.86), 7.506 (4.36), 7.517 (16.00), 7.527 (4.36), 7.828 (1.83), 7.833 (1.66), 7.850 (2.65), 7.855 (2.51 ), 7.924 (5.15), 7.946 (3.20), 8.848 (1.13), 8.863 (1.83), 8.877 (1.05), 12.048 (0.45).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.05 (br. s, 1 H), 8.86 (t, 1 H), 7.93 (d, 1 H), 7.84 (dd, 1 H), 7.67 (br. s, 1 H), 7.55-7.47 (m, 5H), 7.45-7.39 (m, 1 H), 7.35-7.25 (m, 1 H), 7.24-7.13 (m, 2H), 3.83-3.71 (m, 1 H), 3.71 -3.61 (m, 1 H), 3.41 -3.33 (m, 1 H, teilweise verdeckt), 2.20-1.99 (m, 6H), 1.88-1.75 (m, 1 H).
Beispiel 177
(+)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-fluorphenyl)pentansäure (Enantiomer 2)
Bei der in Beispiel 175 beschriebenen Enantiomerentrennung wurden 130 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = +36.9°, 589 nm, c = 0.25 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.00 min; MS (ESIpos): m/z = 535/537 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.1 1 ), 0.008 (1.01 ), 1.786 (0.50), 1.803 (0.70), 1.813 (0.78), 1.825 (0.90), 1.836 (0.61 ), 1.848 (0.49), 2.031 (0.80), 2.043 (0.93), 2.056 (1.99), 2.067 (1.12), 2.073 (1.31 ), 2.084 (3.28), 2.092 (4.41 ), 2.1 15 (3.90), 2.131 (2.64), 2.152 (1.05), 2.523 (0.46), 3.635 (0.44), 3.648 (0.75), 3.667 (0.96), 3.681 (1.32), 3.695 (0.72), 3.733 (0.56), 3.752 (0.80), 3.769 (0.71 ), 7.144 (1.09), 7.165 (1.77), 7.190 (2.21 ), 7.206 (2.04), 7.225 (1.44), 7.271 (0.66), 7.285 (1.15), 7.301 (1.12), 7.322 (0.46), 7.399 (1.13), 7.403 (1.14), 7.418 (2.04), 7.421 (1.92), 7.436 (1.02), 7.480 (0.55), 7.486 (1.15), 7.499 (3.30), 7.506 (3.66), 7.517 (16.00), 7.523 (5.91 ), 7.528 (4.35), 7.828 (1.76), 7.833 (1.58), 7.850 (2.67), 7.855 (2.52), 7.924 (5.27), 7.947 (3.33), 8.849 (1.12), 8.864 (1.90), 8.878 (1.07).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.05 (br. s, 1 H), 8.86 (t, 1 H), 7.93 (d, 1 H), 7.84 (dd, 1 H), 7.67 (br. s, 1 H), 7.54-7.48 (m, 5H), 7.45-7.39 (m, 1 H), 7.36-7.26 (m, 1 H), 7.24-7.12 (m, 2H), 3.82-3.71 (m, 1 H), 3.71 -3.61 (m, 1 H), 3.42-3.33 (m, 1 H, teilweise verdeckt), 2.22-1.98 (m, 6H), 1.89-1.75 (m, 1 H).
Beispiel 178
(+/-)-5-{[(6-Brom-3-methyl-2-phenylchinoN
pentansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 4-[2-(difluormethoxy)phenyl]pentanoat (500 mg, 782 μηηοΙ, Beispiel 253A) in Dichlormethan (5.7 ml) wurde TFA (600 μΙ, 7.8 mmol) gegeben, und das Gemisch wurde über Nacht bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde in Acetonitril aufgenommen und mittels präpa- rativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert. Das Lyophilisat wurde mit Ethylacetat und gesättigter Natriumhydrogen- carbonat-Lösung (jeweils 10 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase zweimal mit Ethylacetat (jeweils 10 ml) extrahiert. Die vereinigten organischen
Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 238 mg (98% Reinheit, 51 % d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.00 min; MS (ESIpos): m/z = 583/585 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.821 (0.58), 1.841 (1.12), 1.862 (1.43), 1.886 (0.90), 2.000 (0.42), 2.022 (1.00), 2.037 (3.07), 2.067 (3.02), 2.077 (4.34), 2.095 (2.55), 2.124 (4.67), 2.146 (2.22), 3.378 (1.23), 3.397 (1.28), 3.636 (0.63), 3.651 (1.02), 3.669 (1.45), 3.684 (1.78), 3.698 (0.98), 3.733 (1.04), 3.751 (1.49), 3.768 (1.39), 3.783 (0.87), 3.803 (0.56), 4.624 (1.03), 6.992 (2.69), 7.158 (3.03), 7.178 (9.00), 7.245 (1.19), 7.263 (3.01 ), 7.281 (2.28), 7.31 1 (2.07), 7.330 (2.45), 7.346 (0.98), 7.363 (2.70), 7.430 (3.02), 7.433 (3.12), 7.448 (2.55), 7.452 (2.49), 7.481 (0.55), 7.494 (1.58), 7.504 (4.68), 7.512 (4.55), 7.523 (15.91 ), 7.525 (16.00), 7.534 (6.68), 7.538 (5.96), 7.680 (0.71 ), 7.838 (2.34), 7.843 (2.22), 7.860 (3.62), 7.865 (3.62), 7.934 (6.72), 7.956 (4.28), 8.832 (1.55), 8.847 (2.98), 8.861 (1.52).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.85 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.68 (br. s, 1 H), 7.56-7.48 (m, 5H), 7.44 (dd, 1 H), 7.38-7.23 (m, 2H), 7.21-6.97 (m, 2H), 3.82-3.72 (m, 1 H), 3.71- 3.60 (m, 1 H), 3.45-3.32 (m, 1 H), 2.21-1.98 (m, 6H), 1.94-1.78 (m, 1 H).
Trennung der Enantiomere:
Die Titelverbindung (230 mg) wurde in Methanol (20 ml) gelöst und mittels praparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 179 und 180 [Säule: Daicel Chiral- pak AD-H, 5 μηι, 250 mm x 20 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur: 40 °C; In- jektion: 1.0 ml; Eluent: 25% Isopropanol / 75% Kohlendioxid; Laufzeit 7 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet.
Beispiel 179
(-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-[2-(difluormethoxy)phenyl]- pentansäure (Enantiomer 1)
Bei der in Beispiel 178 beschriebenen Enantiomerentrennung wurden 60 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -17.7°, 589 nm, c = 0.30 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.03 min; MS (ESIpos): m/z = 583/585 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.52), 0.008 (1.39), 1.030 (4.20), 1.045 (4.27), 1.819 (0.56), 1.840 (1.09), 1.860 (1.40), 1.884 (0.90), 1.999 (0.40), 2.020 (0.94), 2.035 (3.03), 2.065 (2.88), 2.075 (4.17), 2.094 (2.50), 2.123 (4.63), 2.145 (2.12), 3.375 (1.26), 3.394 (1.29), 3.523 (0.73), 3.634 (0.59), 3.648 (0.97), 3.666 (1.40), 3.682 (1.74), 3.696 (0.94), 3.731 (1.00), 3.747 (1.42), 3.766 (1.36), 3.782 (0.83), 3.800 (0.53), 6.991 (2.74), 7.157 (2.98), 7.177 (9.10), 7.243 (1.17), 7.261 (2.98), 7.279 (2.29), 7.310 (2.02), 7.329 (2.40), 7.345 (0.97), 7.363 (2.73),
7.428 (3.01 ), 7.432 (3.03), 7.447 (2.55), 7.451 (2.44), 7.477 (0.57), 7.490 (1.52), 7.500 (4.55), 7.508 (4.30), 7.520 (16.00), 7.530 (6.42), 7.677 (0.70), 7.831 (2.27), 7.836 (2.1 1 ), 7.853 (3.48), 7.859 (3.41 ), 7.929 (6.63), 7.951 (4.24), 8.826 (1.52), 8.841 (2.91 ), 8.855 (1.50), 12.015 (2.69).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.02 (s, 1 H), 8.84 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.68 (br. s, 1 H), 7.56-7.48 (m, 5H), 7.44 (dd, 1 H), 7.38-7.23 (m, 2H), 7.19-6.98 (m, 2H), 3.82- 3.72 (m, 1 H), 3.71-3.61 (m, 1 H), 3.43-3.34 (m, 1 H), 2.20-1.97 (m, 6H), 1.92-1.76 (m, 1 H).
Beispiel 180
(+)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-[2-(difluorm
pentansäure (Enantiomer 2)
Bei der in Beispiel 178 beschriebenen Enantiomerentrennung wurden 67 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = +16.8°, 589 nm, c = 0.33 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.03 min; MS (ESIpos): m/z = 583/585 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.17), 0.008 (1.84), 1.030 (4.30), 1.045 (4.35), 1.819 (0.54), 1.839 (1.03), 1.859 (1.36), 1.883 (0.86), 2.020 (0.90), 2.035 (2.94), 2.075 (4.00), 2.093 (2.37), 2.122 (4.45), 3.374 (1.21 ), 3.394 (1.22), 3.633 (0.56), 3.648 (0.95), 3.666 (1.33), 3.681 (1.65), 3.696 (0.90), 3.730 (0.95), 3.747 (1.42), 3.766 (1.37), 3.779 (0.89), 3.800 (0.51 ), 4.325 (0.54), 4.336 (0.52), 6.991 (2.66), 7.156 (2.92), 7.177 (8.88), 7.243 (1.12), 7.260 (2.91 ), 7.278 (2.22), 7.310 (1.94), 7.328 (2.30), 7.344 (0.94), 7.362 (2.66), 7.428 (2.91 ), 7.431 (2.87), 7.447 (2.49), 7.477 (0.50), 7.490 (1.46), 7.500 (4.46), 7.508 (4.24), 7.520 (16.00), 7.531 (6.21 ), 7.677 (0.71 ), 7.831 (2.33), 7.836 (2.1 1 ), 7.853 (3.53), 7.859 (3.38), 7.928 (6.72), 7.951 (4.28), 8.826 (1.45), 8.841 (2.79), 8.855 (1.41 ), 12.014 (2.07).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.01 (s, 1 H), 8.84 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.68 (br. s, 1 H), 7.56-7.48 (m, 5H), 7.44 (dd, 1 H), 7.38-7.22 (m, 2H), 7.20-6.96 (m, 2H), 3.83- 3.71 (m, 1 H), 3.71-3.60 (m, 1 H), 3.44-3.34 (m, 1 H), 2.20-1.96 (m, 6H), 1.94-1.77 (m, 1 H).
Beispiel 181
(+/-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2,6-difluorphenyl)pentan- säure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 4-(2,6-difluorphenyl)pentanoat (500 mg, 820 μηιοΙ, Beispiel 254A) in Dichlormethan (6.0 ml) wurde TFA (630 μΙ, 8.2 mmol) gegeben, und das Gemisch wurde über Nacht bei RT stehen ge- lassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde in Acetonitril aufgenommen und mittels präparati- ver HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert. Das Lyophilisat wurde mit Ethylacetat und gesättigter Natriumhydrogen- carbonat-Lösung (jeweils 10 ml) versetzt und geschüttelt. Nach Phasentrennung wurde die wässrige Phase zweimal mit Ethylacetat (jeweils 10 ml) extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 442 mg (98% Reinheit, 95% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.92 min; MS (ESIpos): m/z = 553/555 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.64), 0.008 (0.51 ), 1.886 (0.55), 1.901 (0.72), 1.916 (0.93), 1.927 (0.87), 1.958 (0.42), 2.061 (0.90), 2.074 (1.15), 2.092 (1.18), 2.1 12 (1.59), 2.127 (5.66), 2.144 (7.80), 3.521 (0.88), 3.721 (0.74), 3.740 (1.31 ), 3.754 (1.85), 3.769 (1.54), 3.788 (1.01 ), 3.809 (0.81 ), 7.056 (2.54), 7.078 (4.69), 7.101 (3.00), 7.333 (0.88), 7.352 (1.17), 7.369 (0.79), 7.483 (0.41 ), 7.493 (1.20), 7.505 (3.76), 7.512 (3.82), 7.523 (16.00), 7.534 (5.28), 7.842 (2.02), 7.847 (1.84), 7.864 (3.16), 7.869 (3.07), 7.934 (5.90), 7.956 (3.70), 8.942 (1.26), 8.957 (2.42), 8.971 (1.26).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.96 (t, 1 H), 7.94 (d, 1 H), 7.86 (dd, 1 H), 7.71 (br. s, 1 H), 7.56-7.46 (m, 5H), 7.40-7.29 (m, 1 H), 7.08 (t, 2H), 3.86-3.68 (m, 2H), 3.58-3.46 (m, 1 H), 2.22- 2.03 (m, 6H), 1.98-1.85 (m, 1 H).
Trennung der Enantiomere:
Die Titelverbindung (380 mg) wurde in Methanol (20 ml) gelöst und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 182 und 183) [Säule: Daicel Chiral- pak AD, 5 μηη, 250 mm x 20 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injek-
tion: 1.0 ml; Eluent: 25% Isopropanol / 75% Kohlendioxid; Laufzeit 10 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet.
Beispiel 182
(-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2,6-difluorphenyl)pentansäure (Enantiomer 1)
Bei der in Beispiel 181 beschriebenen Enantiomerentrennung wurde die vorgereinigte Titelverbindung als früher eluierendes Enantiomer (ee-Wert 99%) erhalten. Anschließend wurde mittels praparativer HPLC (Methode 14) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Anschließend wurde ein weiteres Mal mittels prä- parativer HPLC (Methode 18) nachgereinigt. Es wurden 27 mg (98% Reinheit) der nachgereinigten Titelverbindung erhalten.
[a]D 20 = -47.5°, 589 nm, c = 0.31 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.92 min; MS (ESIpos): m/z = 553/555 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (3.36), 0.008 (3.27), 1.909 (0.80), 2.054 (0.74), 2.067 (0.89), 2.088 (1.10), 2.114 (4.71 ), 2.130 (4.70), 2.144 (4.68), 2.327 (0.54), 2.670 (0.59), 3.514 (0.89), 3.714 (0.65), 3.733 (1.16), 3.748 (1.58), 3.762 (1.27), 3.782 (0.91 ), 7.054 (2.15), 7.076 (4.00), 7.099 (2.52), 7.332 (0.78), 7.348 (1.00), 7.488 (1.07), 7.501 (3.22), 7.508 (3.48), 7.519 (16.00), 7.835 (1.80), 7.840 (1.66), 7.857 (2.81 ), 7.863 (2.74), 7.928 (5.21 ), 7.951 (3.25), 8.939 (1.07), 8.954 (1.99), 8.969 (1.07).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.00 (br. s, 1 H), 8.95 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.70 (br. s, 1 H), 7.56-7.45 (m, 5H), 7.41-7.25 (m, 1 H), 7.08 (t, 2H), 3.88-3.67 (m, 2H), 3.51 (br. s, 1 H), 2.24-2.01 (m, 6H), 1.99-1.82 (m, 1 H).
Beispiel 183
(+)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2,6-difluorphenyl)pentansäure (Enantiomer 2)
Bei der in Beispiel 181 beschriebenen Enantiomerentrennung wurde die vorgereinigte Titelverbindung als später eluierendes Enantiomer (ee-Wert 99%) erhalten. Anschließend wurde mittels präparativer HPLC (Methode 14) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 56 mg (98% Reinheit) der nachge- reinigten Titelverbindung erhalten.
[a]D 20 = +56.2°, 589 nm, c = 0.38 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 0.99 min; MS (ESIpos): m/z = 553/555 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.882 (0.61 ), 1.897 (0.80), 1.912 (1.00), 1.952 (0.41 ), 2.058 (1.06), 2.073 (2.93), 2.089 (1.45), 2.124 (5.99), 2.140 (7.91 ), 3.517 (0.98), 3.703 (0.47), 3.717 (0.82), 3.735 (1.41 ), 3.750 (1.92), 3.764 (1.59), 3.784 (1.10), 3.804 (0.85), 7.055 (2.46), 7.077 (4.38), 7.100 (2.65), 7.332 (0.99), 7.350 (1.19), 7.367 (0.78), 7.489 (1.84), 7.501 (4.01 ), 7.509 (4.54), 7.519 (16.00), 7.836 (1.87), 7.841 (1.61 ), 7.858 (2.80), 7.863 (2.43), 7.929 (4.66), 7.951 (2.85), 8.935 (1.35), 8.950 (2.28), 8.964 (1.15), 12.082 (1.35).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.08 (br. s, 1 H), 8.95 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.70 (br. s, 1 H), 7.56-7.46 (m, 5H), 7.41 -7.29 (m, 1 H), 7.08 (t, 2H), 3.87-3.67 (m, 2H), 3.52 (br. s, 1 H), 2.21-2.02 (m, 6H), 1.98-1.83 (m, 1 H). Beispiel 184
(+/-)-5-{[(6-Brom-3-methyl-2-phenylcN^
säure (Racemat)
Zu einer Lösung aus (+/-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4- (2-chlor-3-fluorphenyl)pentanoat (100 mg, 160 μηιοΙ, Beispiel 255A) in Dichlormethan (1.2 ml) wurde TFA (120 μΙ, 1.6 mmol) gegeben, und das Gemisch wurde drei Tage bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde in Acetonitril aufgenommen und mittels praparativer HPLC (Methode 15) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert. Es wurden 78 mg (100% Reinheit, 86% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.96 min; MS (ESIpos): m/z = 569/571 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.818 (0.64), 1.838 (1.05), 1.858 (1.38), 1.880 (0.81 ), 2.028 (0.44), 2.049 (0.90), 2.073 (2.99), 2.085 (1.48), 2.098 (3.43), 2.1 10 (5.48), 2.140 (5.75), 2.176 (1.34), 3.631 (1.74), 3.746 (1.83), 7.283 (0.98), 7.304 (2.1 1 ), 7.325 (1.39), 7.364 (1.57), 7.382 (3.21 ), 7.401 (1.38), 7.419 (1.64), 7.434 (1.44), 7.454 (0.55), 7.479 (0.61 ), 7.492 (1.67),
7.502 (4.92), 7.510 (4.75), 7.522 (16.00), 7.533 (6.95), 7.651 (0.43), 7.832 (2.36), 7.837 (2.13), 7.854 (3.59), 7.859 (3.37), 7.930 (6.74), 7.953 (4.29), 8.854 (1.51 ), 8.869 (2.99), 8.883 (1.46).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.09 (br. s, 1 H), 8.87 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.65 (br. s, 1 H), 7.56-7.47 (m, 5H), 7.46-7.35 (m, 2H), 7.34-7.26 (m, 1 H), 3.87-3.35 (m, 3H, teilweise verdeckt), 2.23-2.00 (m, 6H), 1.93-1.75 (m, 1 H).
Beispiel 185
(-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-c
säure (Enantiomer 1)
Zu einer Lösung aus (-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4- (2-chlor-3-fluorphenyl)pentanoat (275 mg, 439 μηηοΙ, Beispiel 256A) in Dichlormethan (3.7 ml) wurde TFA (740 μΙ, 9.7 mmol) gegeben, und das Gemisch wurde 16 h bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde in Acetonitril aufgenommen und mittels praparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rück- stand lyophilisiert. Es wurden 204 mg (100% Reinheit, ee-Wert 96%, 81 % d. Th.) der Titelverbindung erhalten.
[a]D 20 = -18.3°, 589 nm, c = 0.38 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.96 min; MS (ESIpos): m/z = 569/571 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.818 (0.61 ), 1.837 (1.07), 1.857 (1.36), 1.879 (0.84), 2.070 (2.64), 2.084 (1.42), 2.097 (3.39), 2.109 (5.38), 2.140 (5.93), 3.640 (1.33), 3.745 (1.77), 7.283 (1.02), 7.304 (2.13), 7.326 (1.43), 7.364 (1.63), 7.382 (3.24), 7.400 (1.44), 7.419 (1.73), 7.434 (1.48), 7.454 (0.60), 7.491 (1.75), 7.502 (4.64), 7.509 (4.71 ), 7.522 (16.00), 7.643 (0.42), 7.831 (2.09), 7.836 (2.04), 7.853 (3.22), 7.858 (3.21 ), 7.929 (5.64), 7.952 (3.62), 8.853 (1.50), 8.868 (2.89), 8.881 (1.45), 12.068 (0.80).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.07 (br. s, 1 H), 8.87 (t, 1 H), 7.94 (d, 1 H), 7.84 (dd, 1 H), 7.64 (br. s, 1 H), 7.56-7.47 (m, 5H), 7.46-7.35 (m, 2H), 7.34-7.25 (m, 1 H), 3.87-3.55 (m, 3H), 2.22-1.99 (m, 6H), 1.92-1.76 (m, 1 H).
Beispiel 186
(+)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlor-3-fluorphenyl)- pentansäure (Enantiomer 2)
Zu einer Lösung aus (+)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4- (2-chlor-3-fluorphenyl)pentanoat (290 mg, 463 μηηοΙ, Beispiel 257A) in Dichlormethan (3.9 ml) wurde TFA (790 μΙ, 10 mmol) gegeben, und das Gemisch wurde 16 h bei RT stehen gelassen.
Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde in Acetonitril aufgenommen und mittels praparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert. Es wurden 266 mg (100% Reinheit, ee-Wert 98%, 100% d. Th.) der Titelver- bindung erhalten.
[a]D 20 = +18.0°, 589 nm, c = 0.37 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.96 min; MS (ESIpos): m/z = 569/571 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.818 (0.65), 1.838 (1.1 1 ), 1.858 (1.42), 1.880 (0.84), 2.029 (0.40), 2.049 (0.92), 2.071 (2.77), 2.085 (1.40), 2.099 (3.51 ), 2.1 10 (5.62), 2.141 (6.00), 2.176 (1.41 ), 3.518 (2.35), 3.640 (1.85), 7.283 (1.02), 7.304 (2.17), 7.325 (1.44), 7.364 (1.67), 7.382 (3.38), 7.401 (1.44), 7.419 (1.73), 7.434 (1.50), 7.454 (0.57), 7.479 (0.64), 7.492 (1.68), 7.503 (4.84), 7.510 (4.62), 7.522 (16.00), 7.533 (7.15), 7.646 (0.41 ), 7.832 (2.25), 7.837 (2.05), 7.855 (3.46), 7.860 (3.27), 7.930 (6.14), 7.953 (3.94), 8.855 (1.57), 8.870 (3.08), 8.884 (1.51 ).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.10 (br. s, 1 H), 8.87 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.65 (br. s, 1 H), 7.56-7.47 (m, 5H), 7.46-7.34 (m, 2H), 7.34-7.26 (m, 1 H), 3.87-3.58 (m, 3H, teilweise verdeckt), 2.22-2.00 (m, 6H), 1.93-1.76 (m, 1 H).
Beispiel 187
(+/-)-/V-[2-{[(6-Brom-3-methyl-2-phenylc^
methylglycin (Racemat)
Zu einer Lösung aus (+/-)-ie/f-Butyl-/V-[2-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]- amino}-1-(2-chlorphenyl)ethyl]-/V-methylglycinat (150 mg, 241 μηηοΙ, Beispiel 258A) in Dichlormethan (1.8 ml) wurde TFA (190 μΙ, 2.4 mmol) gegeben, und das Gemisch wurde 16 h bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde in Acetonitril aufgenommen und mittels praparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und
der Rückstand lyophilisiert. Anschließend wurde der Rückstand mit Ethylacetat und gesättigter Natriumhydrogencarbonat-Lösung (jeweils 10 ml) versetzt, geschüttelt, und die wässrige Phasen wurde nach Phasentrennung zweimal mit Ethylacetat (10 ml) extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 106 mg (98% Reinheit, 76% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.54 min; MS (ESIpos): m/z = 566/568 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 2.089 (7.35), 2.644 (1.80), 4.062 (0.43), 4.161 (0.56), 5.018 (0.45), 7.434 (1.06), 7.454 (1.84), 7.473 (1.43), 7.513 (16.00), 7.552 (1.61 ), 7.572 (1.18), 7.676 (0.54), 7.761 (0.86), 7.778 (0.78), 7.834 (1.30), 7.839 (1.23), 7.856 (2.01 ), 7.861 (2.01 ), 7.926 (3.68), 7.948 (2.25), 8.881 (1.00).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.88 (br. s, 1 H), 7.93 (d, 1 H), 7.85 (dd, 1 H), 7.81-7.73 (m, 1 H), 7.68 (br. s, 1 H), 7.56 (br. d, 1 H), 7.53-7.40 (m, 7H), 5.02 (br. s, 1 H), 4.12 (br. d, 2H), 3.70 (br. d, 2H), 2.64 (br. s, 3H), 2.09 (s, 3H).
Trennung der Enantiomere:
Die Titelverbindung (70 mg) wurde in Methanol (10 ml) gelöst und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 188 und 189) [Säule: Daicel Chiral- pak AD, 5 μηη, 250 mm x 20 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 1.0 ml; Eluent: 25% Isopropanol / 75% Kohlendioxid; Laufzeit 9 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Beispiel 188
(-)-/V-[2-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-1-(2-chlorphenyl)ethyl]-/V- methylglycin (Enantiomer 1)
Bei der in Beispiel 187 beschriebenen Enantiomerentrennung wurden 22 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als früher eluierendes Enantiomer erhalten. [a]D 20 = -27.9°, 589 nm, c = 0.25 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 0.81 min; MS (ESIpos): m/z = 566/568 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.35), 0.008 (1.09), 2.152 (14.46), 2.327 (0.41 ), 2.362 (16.00), 2.523 (1.06), 3.149 (1.90), 3.191 (2.75), 3.353 (4.75), 3.396 (2.42), 3.827 (0.54), 3.979 (0.65), 4.631 (1.23), 4.648 (1.83), 4.665 (1.1 1 ), 7.306 (0.72), 7.310 (0.81 ), 7.325 (1.95), 7.329 (1.97), 7.344 (1.76), 7.348 (1.69), 7.366 (1.51 ), 7.384 (2.16), 7.400 (0.94), 7.465 (2.88), 7.468 (2.88), 7.485 (3.05), 7.488 (3.02), 7.500 (3.68), 7.507 (3.24), 7.519 (8.83), 7.524 (8.36), 7.532 (5.29), 7.544 (1.15), 7.577 (2.10), 7.581 (2.12), 7.596 (1.80), 7.600 (1.70), 7.742 (1.68), 7.827 (1.80), 7.832 (1.58), 7.849 (2.77), 7.854 (2.62), 7.923 (4.87), 7.945 (3.12), 8.726 (1.02), 8.741 (2.04), 8.755 (0.99).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.19 (br. s, 1 H), 8.74 (t, 1 H), 7.93 (d, 1 H), 7.84 (dd, 1 H), 7.74 (br. s, 1 H), 7.59 (dd, 1 H), 7.56-7.49 (m, 5H), 7.48 (dd, 1 H), 7.38 (td, 1 H), 7.33 (td, 1 H), 4.65 (t, 1 H), 3.98 (br. s, 1 H), 3.83 (br. s, 1 H), 3.37 (d, 1 H, teilweise verdeckt), 3.17 (d, 2H), 2.36 (s, 3H), 2.15 (s, 3H). Beispiel 189
(+)-/V-[2-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amin
methylglycin (Enantiomer 2)
Bei der in Beispiel 187 beschriebenen Enantiomerentrennung wurden 19 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als später eluierendes Enantiomer erhalten. [a]D 20 = +27.1 °, 589 nm, c = 0.25 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 0.81 min; MS (ESIpos): m/z = 566/568 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (1.13), 2.152 (14.59), 2.327 (0.43), 2.362 (16.00), 3.149 (1.93), 3.191 (2.73), 3.353 (4.74), 3.396 (2.45), 3.823 (0.54), 3.979 (0.64), 4.631 (1.21 ), 4.648 (1.81 ), 4.666 (1.16), 7.310 (0.79), 7.325 (1.94), 7.329 (1.97), 7.344 (1.73), 7.348 (1 .72), 7.366 (1.55), 7.385 (2.19), 7.399 (0.90), 7.465 (3.00), 7.468 (3.1 1 ), 7.485 (3.09), 7.488 (3.18), 7.500 (3.65), 7.507 (3.20), 7.519 (8.85), 7.524 (8.42), 7.532 (5.37), 7.580 (2.1 1 ), 7.596 (1.77), 7.742 (1.80), 7.827 (1.91 ), 7.832 (1.64), 7.849 (2.86), 7.854 (2.70), 7.923 (5.10), 7.945 (3.24), 8.727 (1.00), 8.741 (2.02), 8.756 (0.97).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.15 (br. s, 1 H), 8.74 (t, 1 H), 7.93 (d, 1 H), 7.84 (dd, 1 H), 7.74 (br. s, 1 H), 7.59 (dd, 1 H), 7.55-7.49 (m, 5H), 7.48 (dd, 1 H), 7.38 (td, 1 H), 7.33 (td, 1 H), 4.65 (t, 1 H), 3.98 (br. s, 1 H), 3.82 (br. s, 1 H), 3.37 (d, 1 H, teilweise verdeckt), 3.17 (d, 1 H), 2.36 (s, 3H), 2.15 (s, 3H).
Beispiel 190
(+/-)-/V-[2-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-1-(2-chlorphenyl)ethyl]glycin (Racemat)
Zu einer Lösung aus (+/-)-ie/f-Butyl-/V-[2-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]- amino}-1-(2-chlorphenyl)ethyl]glycinat (150 mg, 246 μηηοΙ, Beispiel 259A) in Dichlormethan (1.8 ml) wurde TFA (190 μΙ, 2.5 mmol) gegeben, und das Gemisch wurde 16 h bei RT stehen gelas- sen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde in Acetonitril aufgenommen und mittels praparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert. Anschließend wurde der Rückstand mit Ethylacetat und gesättigter Natrium- hydrogencarbonat-Lösung (jeweils 10 ml) versetzt, geschüttelt, und die wässrige Phasen wurde nach Phasentrennung zweimal mit Ethylacetat (10 ml) extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 52 mg (98% Reinheit, 37% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.47 min; MS (ESIpos): m/z = 552/554 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.150 (0.75), 0.146 (0.72), 1.235 (1.1 1 ), 1.365 (0.81 ), 1.876 (0.72), 2.283 (16.00), 2.327 (2.01 ), 2.366 (1.42), 2.670 (1.90), 2.690 (1.92), 2.709 (1.64), 2.731 (2.26), 2.761 (2.70), 2.802 (2.51 ), 2.890 (2.01 ), 3.569 (1.62), 3.667 (1.39), 4.302 (2.31 ), 7.289 (2.79), 7.306 (2.34), 7.345 (2.23), 7.362 (3.21 ), 7.419 (4.13), 7.438 (3.99), 7.510 (6.94), 7.529 (5.97), 7.567 (7.41 ), 7.672 (2.59), 7.690 (2.51 ), 7.842 (2.51 ), 7.865 (3.43), 7.903 (3.51 ), 7.939 (4.32), 7.960 (2.70), 9.456 (1.23).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 9.46 (br. s, 1 H), 7.99-7.81 (m, 3H), 7.68 (br. d, 1 H), 7.61- 7.25 (m, 9H), 4.30 (br. s, 1 H), 3.67 (br. s, 1 H), 3.54 (br. s, 1 H), 2.84-2.60 (m, 2H), 2.28 (s, 3H).
Beispiel 191
(+/-)-[2-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-1-(2-chlorphenyl)ethoxy]essig- säure (Racemat)
Eine Lösung aus (+/-)-Ethyl-[2-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-1-(2- chlorphenyl)ethoxy]acetat (179 mg, 307 μηηοΙ, Beispiel 260A) in einem Gemisch aus THF (3 ml) und Methanol (1 ml) wurde mit 1 M Natronlauge (1.2 ml, 1.2 mmol) unter Rühren versetzt, und das Gemisch wurde 1.5 h bei RT stehen gelassen. Anschließend wurde das Gemisch mit TFA (97 μΙ, 1.3 mmol) versetzt und direkt (ohne weitere Aufarbeitung) mittels präparativer HPLC (Methode 15) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 164 mg (100% Reinheit, 96% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.97 min; MS (ESIpos): m/z = 553/555 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 2.217 (0.41 ), 2.263 (16.00), 3.910 (3.67), 3.951 (5.90), 4.039 (6.43), 4.080 (4.00), 5.166 (1.86), 5.180 (3.39), 5.194 (1.77), 7.376 (1.03), 7.391 (2.46), 7.395 (2.47), 7.409 (2.68), 7.414 (2.90), 7.421 (2.41 ), 7.437 (2.78), 7.455 (1.35), 7.493 (4.12), 7.496 (4.26), 7.516 (8.14), 7.536 (6.88), 7.558 (7.92), 7.579 (5.24), 7.599 (2.67), 7.824 (2.67), 7.851 (2.45), 7.856 (1.90), 7.874 (3.42), 7.879 (2.91 ), 7.949 (4.95), 7.971 (3.19), 8.946 (1.54), 8.960 (2.80), 8.974 (1.44).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.66 (br. s, 1 H), 8.96 (t, 1 H), 7.96 (d, 1 H), 7.87 (dd, 1 H), 7.82 (br. s, 1 H), 7.63-7.48 (m, 7H), 7.47-7.35 (m, 2H), 5.18 (t, 1 H), 4.06 (d, 1 H), 3.94 (d, 1 H), 3.79 (br. s, 2H), 2.26 (s, 3H).
Beispiel 192
(+)-[2-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-1-(2-chlorphenyl)ethoxy]essig- säure (Enantiomer 1)
Eine Lösung aus (+)-Ethyl-[2-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-1-(2- chlorphenyl)ethoxy]acetat (415 mg, 713 μηηοΙ, Beispiel 261 A) in THF (7.0 ml) und Methanol (2.3 ml) wurde bei RT mit 1 M Natronlauge (2.9 ml, 2.9 mmol) versetzt und 1.5 h bei RT stehen gelassen. Anschließend wurde das Gemisch mit Trifluoressigsäure (230 μΙ, 2.9 mmol) versetzt und direkt (ohne weitere Aufarbeitung) mittels präparativer HPLC (Methode 17) gereinigt. Die
vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 308 mg (100% Reinheit, ee-Wert 99%, 78% d. Th.) der Titelverbindung erhalten.
[a]D 20 = +53.0°, 589 nm, c = 0.46 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.05 min; MS (ESIpos): m/z = 553/555 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.47), 2.263 (16.00), 3.783 (2.00), 3.904 (2.71 ),
3.945 (4.66), 4.035 (5.37), 4.076 (3.05), 5.163 (1.64), 5.177 (3.23), 5.191 (1.60), 7.371 (0.76),
7.375 (0.85), 7.390 (2.20), 7.394 (2.26), 7.408 (2.32), 7.413 (2.47), 7.421 (2.04), 7.437 (2.44), 7.440 (2.62), 7.455 (1.1 1 ), 7.492 (3.62), 7.496 (4.22), 7.514 (7.64), 7.533 (6.13), 7.556 (6.82), 7.560 (5.89), 7.580 (4.34), 7.599 (2.27), 7.603 (2.04), 7.822 (2.29), 7.847 (2.38), 7.853 (1.67), 7.870 (3.34), 7.875 (2.84), 7.946 (5.43), 7.968 (3.50), 8.977 (1.70), 12.693 (2.36).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.69 (s, 1 H), 8.98 (t, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.82 (br. s, 1 H), 7.62-7.47 (m, 7H), 7.47-7.36 (m, 2H), 5.18 (t, 1 H), 4.05 (d, 1 H), 3.93 (d, 1 H), 3.78 (br. s, 2H), 2.26 (s, 3H).
Beispiel 193
(-)-[2-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-1-(2-chlorphenyl)ethoxy]essig- säure (Enantiomer 2)
Eine Lösung aus (-)-Ethyl-[2-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-1-(2- chlorphenyl)ethoxy]acetat (390 mg, 670 μηηοΙ, Beispiel 262A) in THF (6.5 ml) und Methanol (2.2 ml) wurde bei RT mit 1 M Natronlauge (2.7 ml, 2.7 mmol) versetzt und 1.5 h bei RT stehen gelassen. Anschließend wurde das Gemisch mit Trifluoressigsäure (210 μΙ, 2.7 mmol) versetzt und direkt (ohne weitere Aufarbeitung) mittels präparativer HPLC (Methode 17) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 307 mg (100% Reinheit, ee-Wert 99%, 83% d. Th.) der Titelverbindung erhalten.
[a]D 20 = -54.3°, 589 nm, c = 0.44 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.05 min; MS (ESIpos): m/z = 553/555 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (1.08), 2.263 (16.00), 3.783 (1.96), 3.905 (2.66),
3.946 (4.56), 4.036 (5.22), 4.077 (2.98), 5.163 (1.60), 5.177 (3.16), 5.191 (1.54), 7.371 (0.78),
7.376 (0.89), 7.390 (2.23), 7.395 (2.31 ), 7.409 (2.37), 7.414 (2.54), 7.422 (2.07), 7.437 (2.40), 7.440 (2.66), 7.455 (1.10), 7.492 (3.75), 7.496 (4.41 ), 7.514 (7.86), 7.534 (6.17), 7.556 (7.02), 7.560 (6.04), 7.580 (4.26), 7.599 (2.27), 7.603 (2.05), 7.822 (2.33), 7.848 (2.53), 7.853 (1.75), 7.870 (3.52), 7.875 (2.97), 7.946 (5.67), 7.968 (3.63), 8.961 (1.03), 8.975 (1.90), 8.989 (0.98), 12.693 (2.1 1 ).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.69 (br. s, 1 H), 8.97 (t, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.82 (br. s, 1 H), 7.62-7.48 (m, 7H), 7.47-7.36 (m, 2H), 5.18 (t, 1 H), 4.05 (d, 1 H), 3.93 (d, 1 H), 3.78 (br. s, 2H), 2.26 (s, 3H).
Beispiel 194
(+/-)-{[2-{[(6-Brom-3-methyl-2-phenylc^^
sulfanyl}essigsäure (Racemat)
Eine Lösung aus (+/-)-Methyl-{[2-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-1-(2- chlorphenyl)ethyl]sulfanyl}acetat (388 mg, 87% Reinheit, 578 mol, Beispiel 263A) in THF (4.9 ml) und Methanol (2.5 ml) wurde bei RT mit 1 M Natronlauge (3.5 ml, 3.5 mmol) versetzt und 16 h bei RT gerührt. Anschließend wurde das Gemisch direkt (ohne weitere Aufarbeitung) mittels präparativer HPLC (Methode 19) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert. Es wurden 310 mg (98% Reinheit, 92% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.06 min; MS (ESIpos): m/z = 569/571 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 2.073 (0.84), 2.147 (8.51 ), 3.201 (3.59), 3.239 (5.50), 3.357 (5.53), 3.395 (3.63), 3.986 (2.19), 4.816 (1.34), 4.835 (2.38), 4.854 (1.22), 7.306 (0.88), 7.324 (2.07), 7.342 (1.55), 7.385 (1.44), 7.404 (2.33), 7.421 (1.1 1 ), 7.473 (3.71 ), 7.476 (3.66), 7.493 (3.82), 7.496 (4.49), 7.500 (4.38), 7.508 (4.14), 7.518 (16.00), 7.529 (5.23), 7.532 (4.74), 7.609 (2.61 ), 7.625 (2.21 ), 7.777 (0.53), 7.831 (2.28), 7.836 (1.94), 7.853 (3.32), 7.859 (3.04), 7.928 (5.63), 7.951 (3.58), 9.024 (1.27), 9.039 (2.50), 9.053 (1.20).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.65 (br. s, 1 H), 9.04 (t, 1 H), 7.94 (d, 1 H), 7.84 (dd, 1 H), 7.78 (br. s, 1 H), 7.62 (dd, 1 H), 7.55-7.46 (m, 6H), 7.40 (t, 1 H), 7.33 (t, 1 H), 4.84 (t, 1 H), 3.99 (br. s, 2H), 3.37 (d, 1 H), 3.22 (d, 1 H), 2.15 (s, 3H).
Trennunq der Enantiomere:
Die Titelverbindung (250 mg) wurde in Methanol (15 ml) gelöst und mittels präparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 195 und 196) [Säule: Daicel AD-H, 5 μηη, 250 mm x 20 mm; Fluss: 80 ml/min; Detektion: 210 nm; Temperatur: 40 °C; Injektion: 0.5 ml; Eluent: 25% Isopropanol / 75% Kohlendioxid; Laufzeit 13.2 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert.
Beispiel 195
(+)-{[2-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-1-(2-chlorphenyl)ethyl]sulfanyl}^ essigsaure (Enantiomer 1)
Bei der in Beispiel 194 beschriebenen Enantiomerentrennung wurden 1 10 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = +22.8°, 589 nm, c = 0.25 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.97 min; MS (ESIpos): m/z = 569/571 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.70), 1.030 (0.60), 1.046 (0.60), 2.059 (0.59), 2.073 (0.43), 2.145 (8.31 ), 2.523 (0.40), 3.199 (3.42), 3.236 (5.29), 3.353 (5.58), 3.391 (3.45), 3.582 (0.55), 3.983 (1.61 ), 4.812 (1.30), 4.831 (2.31 ), 4.850 (1.17), 7.305 (0.86), 7.322 (2.04), 7.341 (1.53), 7.384 (1.42), 7.402 (2.32), 7.420 (1.13), 7.472 (3.68), 7.475 (3.49), 7.495 (4.93), 7.506 (4.01 ), 7.516 (16.00), 7.527 (5.22), 7.606 (2.57), 7.624 (2.17), 7.773 (0.52), 7.828 (2.15), 7.833 (1.84), 7.850 (3.13), 7.856 (2.89), 7.926 (5.32), 7.948 (3.38), 9.025 (1.18), 9.040 (2.35), 9.054 (1.21 ).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.65 (br. s, 1 H), 9.04 (t, 1 H), 7.93 (d, 1 H), 7.84 (dd, 1 H), 7.77 (br. s, 1 H), 7.62 (d, 1 H), 7.55-7.45 (m, 6H), 7.40 (t, 1 H), 7.33 (t, 1 H), 4.83 (t, 1 H), 3.98 (br. s, 2H), 3.37 (d, 1 H), 3.22 (d, 1 H), 2.15 (s, 3H).
Beispiel 196
(-)-{[2-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-1-(2-chlorphenyl)ethyl]sulfanyl}- essigsäure (Enantiomer 2)
Bei der in Beispiel 194 beschriebenen Enantiomerentrennung wurden 70 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = -25.4°, 589 nm, c = 0.28 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.97 min; MS (ESIpos): m/z = 569/571 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.20), 0.008 (0.96), 1.151 (0.48), 2.058 (0.42), 2.146 (8.57), 2.523 (0.60), 3.194 (3.65), 3.232 (5.76), 3.344 (6.61 ), 3.382 (3.69), 3.980 (1.60),
4.806 (1.31 ), 4.825 (2.35), 4.844 (1.18), 7.304 (0.87), 7.322 (2.08), 7.340 (1.56), 7.383 (1.46), 7.402 (2.36), 7.419 (1.13), 7.471 (3.88), 7.474 (3.73), 7.491 (3.92), 7.495 (4.69), 7.498 (4.45), 7.506 (4.1 1 ), 7.517 (16.00), 7.527 (5.22), 7.530 (4.79), 7.605 (2.60), 7.622 (2.20), 7.776 (0.56), 7.828 (2.40), 7.833 (1.99), 7.850 (3.49), 7.856 (3.13), 7.925 (5.97), 7.948 (3.77), 9.041 (1.14), 9.055 (2.19), 9.069 (1.09).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.69 (br. s, 1 H), 9.06 (t, 1 H), 7.93 (d, 1 H), 7.84 (dd, 1 H), 7.78 (br. s, 1 H), 7.61 (dd, 1 H), 7.56-7.45 (m, 6H), 7.40 (t, 1 H), 7.32 (t, 1 H), 4.83 (t, 1 H), 3.98 (br. s, 2H), 3.36 (d, 1 H), 3.22 (d, 1 H), 2.15 (s, 3H).
Beispiel 197
(+/-)-{[2-{[(6-Brom-3-methyl-2-phen^
sulfonyl}essigsäure (Racemat)
Zu einer Lösung aus (+/-)-Methyl-{[2-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-1- (2-chlorphenyl)ethyl]sulfonyl}acetat (50 mg, 93% Reinheit, 75.5 mol, Beispiel 264A) in THF (740 μΙ) und Methanol (250 μΙ) wurde 1 M Natronlauge (300 μΙ, 300 μηηοΙ) gegeben, und das Gemisch wurde 16 h bei RT stehen gelassen. Anschließend wurde das Gemisch direkt (ohne weitere Aufarbeitung) mittels praparativer HPLC (Methode 17) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 40 mg (98% Reinheit, 86% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.86 min; MS (ESIpos): m/z = 601/603 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 9.23 (br. s, 1 H), 7.92 (d, 1 H), 7.83 (dd, 1 H), 7.76-7.57 (m, 2H), 7.56-7.38 (m, 8H), 5.87 (t, 1 H), 4.42-4.18 (m, 3H), 4.00-3.83 (m, 1 H), 2.01 (br. s, 3H).
Trennung der Enantiomere:
Die Titelverbindung (35 mg) wurde in einem Gemisch aus Ethanol (1 ml) und Dichlormethan (2 ml) gelöst und mittels praparativer HPLC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 198 und 199) [Säule: Daicel Chiralpak IF, 5 μηη, 250 mm x 20 mm; Fluss: 15 ml/min;
Detektion: 220 nm; Temperatur: 35 °C; Injektion: 0.50 ml; Eluent: 60% Heptan / 40% (Ethanol + 0.2% TFA); Laufzeit 12 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert.
Beispiel 198
(+)-{[2-{[(6-Brom-3-methyl-2-phenylchinoN
essigsaure (Enantiomer 1)
Bei der in Beispiel 197 beschriebenen Enantiomerentrennung wurde die vorgereinigte Titelverbindung als früher eluierendes Enantiomer (ee-Wert 99%) erhalten. Anschließend wurde mittels präparativer HPLC (Methode 18) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 23 mg (95% Reinheit, lösungsmit- telhaltig laut 1H-NMR) der nachgereinigten Titelverbindung erhalten.
[a]D 20 = +17.2°, 589 nm, c = 0.25 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.78 min; MS (ESIpos): m/z = 601/603 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 9.32 (s, 1 H), 7.91 (d, 1 H), 7.83 (dd, 1 H), 7.75 (d, 1 H), 7.56-7.37 (m, 9H), 6.02-5.93 (m, 1 H), 4.41-4.15 (m, 2H), 3.91 (d, 1 H), 3.56 (d, 1 H), 2.03 (br. s, 3H).
Beispiel 199
{[2-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-1-(2-chlorphenyl)ethyl]sulfonyl}- essigsäure (Enantiomer 2)
Bei der in Beispiel 197 beschriebenen Enantiomerentrennung wurde die vorgereinigte Titelver- bindung als später eluierendes Enantiomer (ee-Wert 99%) erhalten. Anschließend wurde mittels präparativer HPLC (Methode 18) nachgereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 23 mg (95% Reinheit) der nachgereinigten Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.78 min; MS (ESIpos): m/z = 601/603 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 9.42-9.32 (m, 1 H), 7.91 (d, 1 H), 7.82 (dd, 1 H), 7.75 (d, 1 H), 7.56-7.31 (m, 9H), 5.98 (dd, 1 H), 4.44-4.28 (m, 1 H), 4.26-4.14 (m, 1 H), 3.87 (d, 1 H), 3.52 (d, 1 H), 2.04 (br. s, 3H).
Beispiel 200
(+/-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(6-chlor-2,3-difluorphenyl)- pentansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 4-(6-chlor-2,3-difluorphenyl)pentanoat (121 mg, 188 μηηοΙ, Beispiel 265A) in Dichlormethan (1.6 ml) wurde TFA (320 μΙ, 4.1 mmol) gegeben, und das Gemisch wurde 16 h bei RT stehen gelas- sen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde in Acetonitril aufgenommen und mittels praparativer HPLC (Methode 15) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert. Es wurden 81 mg (100% Reinheit, 73% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.05 min; MS (ESIpos): m/z = 587/589 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (0.62), 1.988 (0.64), 2.073 (0.98), 2.084 (1.18), 2.103 (1.23), 2.121 (2.06), 2.156 (10.15), 2.183 (2.38), 2.206 (0.95), 2.524 (0.67), 3.730 (1.05), 3.766 (0.80), 3.814 (1.72), 7.350 (0.69), 7.373 (2.16), 7.384 (2.42), 7.406 (1.29), 7.426 (1.00), 7.491 (1.49), 7.502 (4.34), 7.510 (4.30), 7.521 (16.00), 7.532 (5.94), 7.838 (2.39), 7.844 (2.04), 7.861 (3.68), 7.866 (3.33), 7.933 (6.34), 7.955 (3.93), 8.940 (1.24), 8.954 (2.38), 8.968 (1.16). 1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.10 (br. s, 1 H), 8.95 (br. t, 1 H), 7.94 (d, 1 H), 7.86 (dd, 1 H), 7.72 (br. s, 1 H), 7.57-7.46 (m, 5H), 7.46-7.32 (m, 2H), 3.96-3.61 (m, 3H), 2.27-1.85 (m, 7H).
Beispiel 201
(-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(6-chlor-2,3-difluorphenyl)- pentansäure (Enantiomer 1)
Zu einer Lösung aus (-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4- (6-chlor-2,3-difluorphenyl)pentanoat (140 mg, 217 μηηοΙ, Beispiel 266A) in Dichlormethan (3.8 ml) wurde TFA (170 μΙ, 2.2 mmol) gegeben, und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde in Acetonitril aufgenommen und mittels präparati- ver HPLC (Methode 14) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der
Rückstand wurde lyophilisiert. Es wurden 93 mg (98% Reinheit, ee-Wert 99%, 71 % d. Th.) der Titelverbindung erhalten.
[a]D 20 = -31.6°, 589 nm, c = 0.25 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.08 min; MS (ESIpos): m/z = 587/569 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.991 (0.63), 2.071 (0.93), 2.083 (1.12), 2.102 (1.18), 2.1 18 (1.94), 2.156 (10.24), 2.179 (2.39), 3.730 (1.04), 3.766 (0.82), 3.813 (1.70), 7.372 (2.13), 7.384 (2.40), 7.405 (1.29), 7.426 (1.00), 7.491 (1.42), 7.502 (4.04), 7.509 (3.99), 7.521 (16.00), 7.838 (2.18), 7.843 (2.00), 7.861 (3.31 ), 7.866 (3.21 ), 7.932 (5.89), 7.955 (3.64), 8.940 (1.16), 8.955 (2.24), 8.969 (1.13).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.1 1 (br. s, 1 H), 8.96 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.72 (br. s, 1 H), 7.55-7.46 (m, 5H), 7.46-7.33 (m, 2H), 3.92-3.63 (m, 3H), 2.28-1.82 (m, 7H).
Beispiel 202
(+)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(6-chlor-2,3-difluorphenyl)- pentansäure (Enantiomer 2)
Zu einer Lösung aus (+)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4- (6-chlor-2,3-difluorphenyl)pentanoat (145 mg, 225 μηηοΙ, Beispiel 267A) in Dichlormethan (4.0 ml) wurde TFA (170 μΙ, 2.3 mmol) gegeben, und das Gemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde in Acetonitril aufgenommen und mittels präparati- ver HPLC (Methode 14) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 94 mg (98% Reinheit, ee-Wert 93%, 70% d. Th.) der Titelverbindung erhalten.
[a]D 20 = +34.9°, 589 nm, c = 0.28 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.08 min; MS (ESIpos): m/z = 587/589 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.153 (0.91 ), 1.180 (0.64), 1.992 (0.63), 2.074 (0.95), 2.087 (1.14), 2.105 (1.16), 2.125 (1.89), 2.156 (10.43), 2.188 (2.43), 2.210 (0.95), 3.731 (1.05), 3.768 (0.82), 3.815 (1.72), 7.373 (2.18), 7.385 (2.46), 7.406 (1.30), 7.428 (1.04), 7.492 (1.55), 7.503 (4.33), 7.510 (4.32), 7.522 (16.00), 7.532 (6.01 ), 7.840 (2.40), 7.845 (2.15), 7.862 (3.65), 7.867 (3.48), 7.934 (6.44), 7.956 (3.98), 8.940 (1.22), 8.955 (2.34), 8.969 (1.15).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.08 (br. s, 1 H), 8.95 (t, 1 H), 7.94 (d, 1 H), 7.86 (dd, 1 H), 7.72 (br. s, 1 H), 7.55-7.47 (m, 5H), 7.45-7.33 (m, 2H), 3.93-3.62 (m, 3H), 2.28-1.87 (m, 7H).
Beispiel 203
(+/-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amin
pentansäure (Racemat)
Zu einer Lösung aus (+/-)-Methyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4- [2-(trifluormethyl)phenyl]pentanoat (500 mg, 96% Reinheit, 801 mol, Beispiel 268A) in THF (7.8 ml) und Methanol (2.6 ml) wurde 1 M Natronlauge (3.2 ml, 3.2 mmol) gegeben, und das Gemisch wurde 16 h bei RT stehen gelassen. Anschließend wurde das Gemisch direkt (ohne weitere Aufarbeitung) mittels präparativer HPLC (Methode 17) gereinigt. Die vereinigten Zielfrak- tionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 415 mg (98% Reinheit, 87% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.09 min; MS (ESIpos): m/z = 585/587 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (3.38), 0.008 (2.76), 1.667 (1.46), 1.681 (1.85), 1.691 (2.53), 1.714 (2.17), 1.725 (1.83), 1.758 (0.78), 1.780 (1.28), 1.820 (5.23), 1.832 (2.78), 1.844 (2.63), 1.856 (1.82), 1.879 (0.92), 1.968 (1.06), 1.982 (1.59), 1.996 (1.85), 2.014 (1.74), 2.026 (2.03), 2.179 (5.86), 2.258 (0.88), 2.328 (0.58), 2.366 (0.51 ), 2.670 (0.56), 2.710 (0.46), 3.606 (1.44), 3.622 (1.90), 3.638 (2.00), 3.719 (1.35), 3.735 (2.21 ), 3.751 (2.00), 3.767 (1.50), 7.426 (1.77), 7.440 (3.06), 7.458 (2.01 ), 7.472 (1.21 ), 7.476 (1.17), 7.488 (3.64), 7.499 (5.24), 7.504 (1 1.48), 7.509 (8.35), 7.523 (13.20), 7.542 (16.00), 7.561 (4.59), 7.661 (1.58), 7.681 (7.35), 7.694 (15.24), 7.710 (6.12), 7.826 (4.03), 7.831 (3.77), 7.849 (6.03), 7.854 (5.92), 7.929 (10.87), 7.952 (7.22), 9.401 (2.02).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 9.40 (br. s, 1 H), 7.94 (d, 1 H), 7.84 (dd, 1 H), 7.80-7.58 (m, 4H), 7.57-7.39 (m, 6H), 3.81-3.69 (m, 1 H), 3.68-3.55 (m, 1 H), 3.37-3.24 (m, 1 H, verdeckt), 2.18 (br. s, 3H), 2.06-1.94 (m, 1 H), 1.91-1.75 (m, 2H), 1.74-1.60 (m, 1 H).
Trennunq der Enantiomere:
Die Titelverbindung (380 mg) wurde in Methanol (30 ml) aufgenommen, filtriert und mittels prä- parativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 204 und 205) [Säule: Daicel Chiralcel AD-H, 5 μηη, 250 mm x 20 mm; Fluss: 80 ml/min; Injektion: 0.50 ml; Eluent: 25% Isopropanol / 75% Kohlendioxid; Laufzeit 6 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert.
Beispiel 204
(-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-[2-(trifluormethyl)phenyl]- pentansäure (Enantiomer 1)
Bei der in Beispiel 203 beschriebenen Enantiomerentrennung wurden 122 mg (98% Reinheit, ee-Wert 99%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -21.3°, 589 nm, c = 0.29 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.1 1 min; MS (ESIpos): m/z = 585/587 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.59), 0.008 (1.00), 1.030 (1.73), 1.046 (1.73), 1.876 (2.12), 1.889 (2.92), 1.904 (2.98), 1.926 (2.60), 1.983 (1.27), 2.004 (2.14), 2.027 (2.52), 2.042 (1.42), 2.063 (2.62), 2.084 (1.71 ), 2.097 (1.99), 2.105 (2.02), 2.164 (4.49), 2.248 (0.63), 2.328 (0.41 ), 2.524 (1.71 ), 2.670 (0.40), 3.625 (1.75), 3.643 (2.03), 3.657 (2.01 ), 3.762 (2.00), 3.778 (1.81 ), 3.794 (1.41 ), 7.446 (1.79), 7.465 (3.56), 7.488 (4.35), 7.499 (5.24), 7.504 (8.90), 7.510 (7.19), 7.523 (13.1 1 ), 7.536 (16.00), 7.555 (3.03), 7.624 (0.60), 7.678 (2.04), 7.698 (4.60), 7.71 1 (6.35), 7.729 (8.88), 7.748 (2.18), 7.830 (3.68), 7.836 (3.32), 7.853 (5.27), 7.858 (4.94), 7.934 (9.75), 7.956 (6.33), 8.991 (2.64).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.99 (br. t, 1 H), 7.94 (d, 1 H), 7.84 (dd, 1 H), 7.78-7.57 (m, 4H), 7.57-7.42 (m, 6H), 3.85-3.72 (m, 1 H), 3.70-3.59 (m, 1 H), 3.37-3.27 (m, 1 H, teilweise verdeckt), 2.23-1.96 (m, 5H), 1.95-1.83 (m, 2H). Beispiel 205
(+)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-[2-(trifluormethyl)phenyl]- pentansäure (Enantiomer 2)
Bei der in Beispiel 203 beschriebenen Enantiomerentrennung wurden 109 mg (98% Reinheit, ee-Wert 98%) der Titelverbindung als später eluierendes Enantiomer erhalten. [a]D 20 = +22.7°, 589 nm, c = 0.37 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.1 1 min; MS (ESIpos): m/z = 585/587 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.81 ), 1.031 (3.90), 1.046 (3.97), 1.858 (0.66), 1.889 (1.78), 1.909 (2.85), 1.926 (2.61 ), 1.938 (1.68), 1.949 (2.14), 2.004 (1.36), 2.023 (2.06), 2.046 (2.38), 2.062 (1.51 ), 2.080 (2.49), 2.109 (1.99), 2.1 18 (2.20), 2.166 (4.52), 2.250 (0.57), 3.631 (1.49), 3.648 (1.82), 3.664 (1.87), 3.766 (1.88), 3.774 (1.80), 3.782 (1.72), 3.799 (1.33), 7.450 (1.66), 7.469 (3.74), 7.489 (4.62), 7.505 (8.38), 7.51 1 (6.77), 7.524 (12.77), 7.537 (16.00), 7.555 (3.12), 7.629 (0.55), 7.682 (1.86), 7.701 (4.31 ), 7.715 (6.05), 7.734 (8.49), 7.755 (2.27), 7.832 (3.52), 7.837 (3.31 ), 7.854 (5.18), 7.859 (5.12), 7.935 (9.70), 7.957 (6.31 ), 8.942 (1.72), 8.955 (3.21 ), 8.969 (1.68).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 8.96 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.78-7.58 (m, 4H), 7.57-7.43 (m, 6H), 3.85-3.72 (m, 1 H), 3.71 -3.59 (m, 1 H), 3.38-3.29 (m, 1 H), 2.23-1.99 (m, 5H), 1.98-1.82 (m, 2H).
Beispiel 206
(+/-)-5-{[(6-Brom-3-methyl-2-phenylch^
pentansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 4-(5-fluor-2-methylphenyl)pentanoat (122 mg, 202 μηιοΙ, Beispiel 269A) in Dichlormethan (1.7 ml) wurde TFA (340 μΙ, 4.4 mmol) gegeben, und das Gemisch wurde 16 h bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde in Acetonitril aufgenommen und mittels praparativer HPLC (Methode 15) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt und der Rückstand lyophilisiert. Es wurden 91 mg (100% Reinheit, 82% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.05 min; MS (ESIpos): m/z = 549/551 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.76), 0.969 (0.40), 1.773 (0.79), 1.788 (0.99), 1.796 (1.02), 1.807 (1.15), 1.812 (1.15), 1.830 (0.89), 1.845 (0.48), 1.981 (0.45), 2.001 (1.09), 2.013 (1.39), 2.032 (1.49), 2.051 (1.65), 2.080 (5.67), 2.096 (8.09), 2.1 12 (4.66), 2.295 (14.18),
2.524 (0.71 ), 3.505 (0.93), 3.518 (1.31 ), 3.538 (1.41 ), 3.552 (1.61 ), 3.565 (0.95), 3.724 (0.98), 3.743 (1.45), 3.762 (1.27), 3.776 (1.15), 3.796 (0.69), 6.920 (0.96), 6.941 (1.82), 6.957 (1.03), 7.156 (2.15), 7.162 (2.21 ), 7.183 (2.42), 7.190 (4.02), 7.207 (2.52), 7.21 1 (2.39), 7.227 (1.87), 7.478 (0.56), 7.491 (1.66), 7.501 (4.98), 7.509 (4.71 ), 7.520 (16.00), 7.531 (7.07), 7.648 (0.56), 7.833 (2.49), 7.838 (2.35), 7.855 (3.81 ), 7.860 (3.73), 7.932 (6.89), 7.954 (4.42), 8.836 (1.61 ), 8.852 (2.38), 8.865 (1.56).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.06 (br. s, 1 H), 8.85 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.65 (br. s, 1 H), 7.56-7.45 (m, 5H), 7.24-7.13 (m, 2H), 6.94 (td, 1 H), 3.76 (td, 1 H), 3.54 (td, 1 H), 3.42-3.30 (1 H, verdeckt), 2.30 (s, 3H), 2.16-1.96 (m, 6H), 1.87-1.73 (m, 1 H). Beispiel 207
(-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(5^
säure (Enantiomer 1)
Zu einer Lösung aus (-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4- (5-fluor-2-methylphenyl)pentanoat (1 12 mg, 185 μηηοΙ, Beispiel 270A) in Dichlormethan (1.6 ml) wurde TFA (310 μΙ, 4.1 mmol) gegeben, und das Gemisch wurde über Nacht bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 17) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 95 mg (100% Reinheit, ee-Wert 99%, 93% d. Th.) der Titelverbindung erhalten. [a]D 20 = -20.0°, 589 nm, c = 0.34 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.98 min; MS (ESIpos): m/z = 549/551 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (1.08), 1.769 (0.75), 1.784 (1.00), 1.792 (1.01 ), 1.807 (1.16), 1.822 (0.89), 1.841 (0.49), 1.976 (0.49), 1.996 (1.09), 2.009 (1.38), 2.028 (1.46), 2.036 (1.15), 2.046 (1.73), 2.073 (5.26), 2.089 (7.14), 2.105 (5.18), 2.294 (14.25), 3.503 (0.90), 3.516 (1.28), 3.537 (1.37), 3.550 (1.61 ), 3.564 (0.94), 3.724 (0.98), 3.742 (1.45), 3.762 (1.25), 3.775 (1.14), 3.796 (0.69), 6.918 (0.95), 6.939 (1.84), 6.954 (1.05), 7.154 (2.15), 7.160 (2.16), 7.181 (2.40), 7.188 (3.89), 7.205 (2.56), 7.209 (2.38), 7.226 (1.89), 7.477 (0.54), 7.490 (1.62), 7.500 (4.98), 7.508 (4.63), 7.520 (16.00), 7.531 (7.15), 7.647 (0.58), 7.832 (2.54), 7.837 (2.31 ), 7.854 (3.87), 7.859 (3.68), 7.931 (6.92), 7.953 (4.45), 8.838 (1.58), 8.853 (2.39), 8.866 (1.54). 1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.1 1 (br. s, 1 H), 8.85 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.65 (br. s, 1 H), 7.56-7.44 (m, 5H), 7.25-7.1 1 (m, 2H), 6.94 (td, 1 H), 3.82-3.69 (m, 1 H), 3.59-3.48 (m, 1 H), 3.4-3.3 (1 H, verdeckt), 2.29 (s, 3H), 2.18-1.95 (m, 6H), 1.86-1.73 (m, 1 H).
Beispiel 208
(+)-5-{[(6-Brom-3-methyl-2-phenylcN^
pentansäure (Enantiomer 2)
Zu einer Lösung aus (+)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4- (5-fluor-2-methylphenyl)pentanoat (1 10 mg, 182 μηιοΙ, Beispiel 271A) in Dichlormethan (1.5 ml) wurde TFA (310 μΙ, 4.0 mmol) gegeben, und das Gemisch wurde über Nacht bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 17) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wur- den 93 mg (100% Reinheit, ee-Wert 97%, 93% d. Th.) der Titelverbindung erhalten.
[a]D 20 = +16.3°, 589 nm, c = 0.37 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.98 min; MS (ESIpos): m/z = 549/551 [M+H]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.069 (0.42), 1.771 (0.79), 1.787 (1.01 ), 1.794 (1.04), 1.805 (1.19), 1.825 (0.92), 1.843 (0.50), 1.979 (0.49), 1.998 (1.12), 2.012 (1.42), 2.030 (1.52), 2.049 (1.74), 2.077 (5.56), 2.093 (7.82), 2.109 (5.15), 2.294 (14.31 ), 3.504 (0.87), 3.517 (1.27), 3.537 (1.36), 3.551 (1.60), 3.565 (0.93), 3.724 (0.97), 3.742 (1.47), 3.761 (1.28), 3.775 (1.15), 3.796 (0.67), 6.919 (1.00), 6.939 (1.89), 6.956 (1.07), 7.155 (2.18), 7.161 (2.23), 7.189 (4.00), 7.206 (2.58), 7.226 (1.83), 7.476 (0.63), 7.490 (1.71 ), 7.500 (5.00), 7.508 (4.64), 7.520 (16.00), 7.647 (0.60), 7.832 (2.38), 7.837 (2.20), 7.854 (3.59), 7.859 (3.49), 7.931 (6.51 ), 7.953 (4.17), 8.837 (1.60), 8.851 (2.45), 8.865 (1.55).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.08 (br. s, 1 H), 8.85 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.65 (br. s, 1 H), 7.56-7.45 (m, 5H), 7.25-7.13 (m, 2H), 6.94 (td, 1 H), 3.82-3.68 (m, 1 H), 3.60-3.48 (m, 1 H), 3.4-3.3 (1 H, verdeckt), 2.29 (s, 3H), 2.19-1.95 (m, 6H), 1.86-1.71 (m, 1 H).
Beispiel 209
(+/-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-[2-(trifluormethoxy)phenyl]- pentansäure (Racemate)
Zu einer Lösung aus (+/-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4- [2-(trifluormethoxy)phenyl]pentanoat (250 mg, 380 μηηοΙ, Beispiel 272A) in Dichlormethan (5.0 ml) wurde TFA (290 μΙ, 3.8 mmol) gegeben, und das Gemisch wurde über Nacht bei RT gerührt. An- schließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde in Acetonitril aufgenommen und mittels präparativer HPLC (Methode 14) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 192 mg (98% Reinheit, 82% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.15 min; MS (ESIpos): m/z = 601/603 [M+H]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.826 (1.37), 1.836 (1.46), 1.847 (1.91 ), 1.870 (1.28), 2.054 (2.96), 2.062 (2.63), 2.082 (10.68), 2.109 (2.89), 2.146 (4.05), 2.525 (1.39), 3.412 (2.61 ), 3.627 (0.95), 3.642 (1.39), 3.660 (2.20), 3.676 (2.63), 3.691 (1.67), 3.703 (1.63), 3.720 (2.17), 3.738 (1.79), 3.754 (1.04), 3.772 (0.61 ), 7.341 (2.05), 7.359 (3.90), 7.381 (1.83), 7.400 (5.00), 7.409 (4.18), 7.418 (4.79), 7.437 (1.32), 7.478 (1.54), 7.490 (2.82), 7.494 (2.85), 7.504 (7.28), 7.51 1 (6.65), 7.523 (16.00), 7.529 (14.55), 7.536 (9.55), 7.548 (2.33), 7.565 (3.86), 7.581 (2.94), 7.588 (2.71 ), 7.655 (0.64), 7.835 (3.21 ), 7.841 (2.87), 7.858 (4.67), 7.863 (4.30), 7.934 (8.58), 7.957 (5.43), 8.864 (2.03), 8.879 (3.53), 8.893 (1.77).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.07 (br. s, 1 H), 8.88 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.66 (br. s, 1 H), 7.60-7.46 (m, 6H), 7.45-7.31 (m, 3H), 3.86-3.56 (m, 2H), 3.48-3.33 (m, 1 H, teilweise verdeckt), 2.25-1.97 (m, 6H), 1.91-1.76 (m, 1 H).
Beispiel 210
(+)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-[2-(trifluormethoxy)phenyl]- pentansäure (Enantiomer 1)
Zu einer Lösung aus (+)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4- [2-(trifluormethoxy)phenyl]pentanoat (1.00 g, 1.52 mmol, Beispiel 273A) in Dichlormethan (10 ml) wurde TFA (1.2 ml, 15 mmol) gegeben, und das Gemisch wurde über Nacht bei RT stehen ge-
lassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 19) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 782 mg (98% Reinheit, 94% d. Th.) der Titelverbindung erhalten. [a]D 20 = +20.8°, 589 nm, c = 0.30 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.08 min; MS (ESIpos): m/z = 601/603 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.23), 0.008 (1.09), 1.827 (1.38), 1.838 (1.41 ), 1.849 (1.97), 1.872 (1.34), 2.058 (2.52), 2.074 (4.27), 2.086 (1 1.66), 2.1 12 (2.61 ), 2.146 (3.88), 2.333 (0.49), 3.415 (1.70), 3.429 (1.69), 3.628 (1.61 ), 3.643 (2.22), 3.661 (3.30), 3.677 (3.96), 3.692 (3.02), 3.703 (3.03), 3.720 (3.62), 3.738 (3.22), 3.754 (2.28), 3.772 (1.71 ), 7.341 (1.76), 7.360 (3.77), 7.381 (1.51 ), 7.400 (4.95), 7.410 (4.07), 7.419 (4.86), 7.437 (1.15), 7.479 (0.88), 7.490 (2.03), 7.504 (6.72), 7.51 1 (5.87), 7.524 (16.00), 7.529 (14.91 ), 7.536 (9.93), 7.548 (2.26), 7.566 (3.87), 7.582 (3.03), 7.589 (2.94), 7.658 (0.62), 7.837 (3.15), 7.842 (2.93), 7.859 (4.76), 7.864 (4.68), 7.935 (9.45), 7.958 (6.02), 8.862 (1.93), 8.877 (3.73), 8.891 (1.94).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.1 1 (br. s, 1 H), 8.88 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.66 (br. s, 1 H), 7.60-7.47 (m, 6H), 7.45-7.31 (m, 3H), 3.79-3.61 (m, 2H), 3.47-3.37 (m, 1 H), 2.24-1.99 (m, 6H), 1.92-1.75 (m, 1 H).
Beispiel 211
(-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-[2-(trifluormethoxy)phenyl]- pentansäure (Enantiomer 2)
Zu einer Lösung aus (-)-ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4- [2-(trifluormethoxy)phenyl]pentanoat (1.00 g, 1.52 mmol, Beispiel 274A) in Dichlormethan (9.4 ml) wurde TFA (1.2 ml, 15 mmol) gegeben, und das Gemisch wurde über Nacht bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 19) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 726 mg (98% Reinheit, 78% d. Th.) der Titelverbindung erhalten.
[a]D 20 = -18.6°, 589 nm, c = 0.34 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.10 min; MS (ESIpos): m/z = 601/603 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.09), 0.008 (1.21 ), 1.814 (0.89), 1.825 (1.42), 1.837 (1.47), 1.848 (1.95), 1.871 (1.29), 2.019 (0.40), 2.058 (2.85), 2.063 (2.58), 2.085 (1 1.46), 2.1 12 (2.86), 2.145 (3.97), 3.627 (0.89), 3.642 (1.34), 3.661 (2.18), 3.676 (2.64), 3.691 (1.66), 3.702 (1.64), 3.719 (2.17), 3.737 (1.80), 3.752 (1.00), 3.771 (0.61 ), 7.341 (1.92), 7.356 (3.21 ), 7.360 (3.85), 7.363 (3.12), 7.381 (1.75), 7.400 (4.97), 7.409 (4.13), 7.419 (4.77), 7.437 (1.16),
7.478 (1.32), 7.490 (2.64), 7.493 (2.63), 7.504 (7.12), 7.51 1 (6.61 ), 7.523 (16.00), 7.528 (14.76), 7.536 (9.48), 7.548 (2.27), 7.565 (3.91 ), 7.582 (2.98), 7.589 (2.80), 7.652 (0.61 ), 7.836 (3.23), 7.841 (2.90), 7.858 (4.74), 7.864 (4.41 ), 7.934 (8.93), 7.957 (5.65), 8.861 (2.07), 8.875 (3.64), 8.889 (1.81 ).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.06 (br. s, 1 H), 8.88 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.65 (br. s, 1 H), 7.61 -7.47 (m, 6H), 7.45-7.31 (m, 3H), 3.80-3.60 (m, 2H), 3.5-3.4 (1 H, verdeckt), 2.24-2.00 (m, 6H), 1.91-1.76 (m, 1 H).
Beispiel 212
(+/-)-5-{[(6-Brom-3-chlor-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-ch
(Racemat)
Zu einer Lösung aus (+/-)-ie/f-Butyl-5-{[(6-brom-3-chlor-2-phenylchinolin-4-yl)carbonyl]amino}-4- (2-chlorphenyl)pentanoat (150 mg, 239 μηηοΙ, Beispiel 275A) in Dichlormethan (2.0 ml) wurde TFA (370 μΙ, 4.8 mmol) gegeben, und das Gemisch wurde über Nacht bei RT gerührt. Anschlie- ßend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 14) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 100 mg (100% Reinheit, 73% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.08 min; MS (ESIpos): m/z = 571/573/575 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 9.13 (br. s, 1 H), 8.01 (d, 1 H), 7.96 (dd, 1 H), 7.70-7.63 (m, 3H), 7.58-7.52 (m, 3H), 7.47 (dd, 2H), 7.37 (t, 1 H), 7.31 -7.20 (m, 1 H), 3.80-3.52 (m, 3H), 2.15-1.92 (m, 3H), 1.90-1.77 (m, 1 H).
Beispiel 213
(-)-5-{[(6-Brom-3-chlor-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlorphenyl)pentansäure (Enantiomer 1)
Zu einer Lösung aus (-)-ie/f-Butyl-5-{[(6-brom-3-chlor-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2- chlorphenyl)pentanoat (715 mg, 1.14 mmol, Beispiel 276A) in Dichlormethan (8.8 ml) wurde TFA (1.9 ml, 25 mmol) gegeben, und das Gemisch wurde 22 h bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder ein- geengt. Der Rückstand wurde in 6 ml Acetonitril gelöst. 2 ml dieser entstandenen Rohprodukt- Lösung wurden mittels präparativer HPLC (Methode 17) gereinigt, und die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 337 mg (100% Reinheit, 52% d. Th., siehe Analytik) einer ersten Fraktion der Titelverbindung erhalten. Aus der verbliebenen Rohprodukt-Lösung (4 ml) fiel ein Feststoff aus, der auch nach Zugabe von weiteren 2 ml Acetonitril nicht mehr in Lösung ging. Nach 3 h Stehen bei RT wurde daher der Feststoff abfiltriert, einmal mit Acetonitril (1 ml) gewaschen und im Vakuum getrocknet. Es wurden 197 mg (100% Reinheit, 30% d. Th.) einer zweiten Charge der Titelverbindung erhalten.
[a]D 20 = -10.6°, 589 nm, c = 0.36 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.09 min; MS (ESIpos): m/z = 571/573/575 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.365 (0.80), 1.832 (1.80), 1.857 (2.81 ), 1.880 (1.60), 1.901 (0.60), 2.050 (1.74), 2.063 (3.99), 2.074 (3.56), 2.090 (7.57), 2.1 14 (4.60), 2.123 (3.87), 2.149 (2.58), 2.171 (1.21 ), 3.615 (2.49), 3.714 (4.45), 3.726 (5.85), 7.257 (2.21 ), 7.275 (5.13), 7.293 (3.66), 7.360 (3.07), 7.379 (5.51 ), 7.397 (2.83), 7.450 (6.62), 7.470 (5.52), 7.498 (5.87), 7.520 (5.94), 7.530 (14.76), 7.536 (16.00), 7.545 (12.64), 7.662 (9.54), 7.667 (1 1.41 ), 7.676 (12.92), 7.942 (3.25), 7.947 (3.02), 7.965 (6.62), 7.969 (6.33), 8.006 (10.46), 8.028 (5.08), 8.991 (2.69), 9.005 (5.23), 9.019 (2.56), 12.041 (0.92).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.04 (br. s, 1 H), 9.00 (t, 1 H), 8.01 (d, 1 H), 7.96 (dd, 1 H), 7.71-7.64 (m, 3H), 7.59-7.43 (m, 5H), 7.38 (t, 1 H), 7.28 (t, 1 H), 3.81 -3.66 (m, 2H), 3.65-3.54 (m, 1 H), 2.22-1.99 (m, 3H), 1.93-1.75 (m, 1 H). Beispiel 214
(+)-5-{[(6-Brom-3-chlor-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlorphenyl)pentansäure (Enantiomer 2)
Zu einer Lösung aus (+)-ie/f-Butyl-5-{[(6-brom-3-chlor-2-phenylchinolin-4-yl)carbonyl]amino}-4- (2-chlorphenyl)pentanoat (735 mg, 1.17 mmol, Beispiel 277A) in Dichlormethan (9.0 ml) wurde TFA (2.0 ml, 26 mmol) gegeben, und das Gemisch wurde 22 h bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde in 8 ml Acetonitril aufgenommen. Dieses Gemisch wurde in der Wärme mit Ultraschall behandelt und anschließend 3 h bei RT stehen gelassen. Der vorhandene Feststoff wurde abfiltriert, zweimal mit Acetonitril (jeweils 1 ml) gewaschen und im Vakuum ge- trocknet. Es wurden 522 mg (100% Reinheit, 78% d. Th.) der Titelverbindung erhalten.
[a]D 20 = +1 1.7°, 589 nm, c = 0.53 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.09 min; MS (ESIpos): m/z = 571/573/575 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.365 (0.85), 1.832 (1.62), 1.847 (1.64), 1.858 (2.62), 1.880 (1.50), 1.901 (0.55), 2.050 (1.57), 2.063 (3.64), 2.074 (2.95), 2.080 (2.51 ), 2.090 (6.93), 2.100 (4.42), 2.1 14 (4.05), 2.123 (3.57), 2.149 (2.48), 2.159 (1.28), 2.171 (1.18), 3.616 (2.16), 3.713 (3.90), 3.726 (5.23), 7.255 (1.92), 7.258 (2.17), 7.277 (4.93), 7.293 (3.39), 7.296 (3.52), 7.361 (2.97), 7.380 (5.38), 7.398 (2.68), 7.449 (6.16), 7.452 (6.60), 7.469 (5.16), 7.472 (5.38), 7.500 (5.52), 7.520 (5.31 ), 7.530 (14.83), 7.536 (16.00), 7.547 (12.13), 7.555 (3.66), 7.571 (0.74), 7.653 (1.69), 7.661 (8.93), 7.667 (10.86), 7.676 (12.18), 7.685 (7.49), 7.942 (3.44), 7.947 (3.32), 7.965 (6.91 ), 7.970 (7.12), 8.006 (1 1.94), 8.028 (5.78), 8.990 (2.47), 9.005 (5.10), 9.019 (2.45), 12.042 (2.44).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.04 (br. s, 1 H), 9.00 (t, 1 H), 8.01 (d, 1 H), 7.96 (dd, 1 H), 7.72-7.63 (m, 3H), 7.58-7.43 (m, 5H), 7.38 (t, 1 H), 7.31 -7.23 (m, 1 H), 3.80-3.67 (m, 2H), 3.66-3.56 (m, 1 H), 2.20-1.98 (m, 3H), 1.92-1.78 (m, 1 H). Beispiel 215
(+/-)-5-{[(6-Brom-3-chlor-2-phenylchinolin-4-yl)carbonyl]amino}-4-[2-(trifluormethyl)phenyl]- pentansäure (Racemat)
Zu einer Lösung aus (+/-)-ie/f-Butyl-5-{[(6-brom-3-chlor-2-phenylchinolin-4-yl)carbonyl]amino}-4- [2-(trifluormethyl)phenyl]pentanoat (80 mg, 121 μηιοΙ, Beispiel 280A) in Dichlormethan (1.0 ml) wurde TFA (200 μΙ, 2.7 mmol) gegeben, und das Gemisch wurde 3.5 h bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 17) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 57 mg (100% Reinheit, 78% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.13 min; MS (ESIpos): m/z = 605/607 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.917 (1.64), 1.926 (1.70), 1.939 (3.77), 1.953 (2.29), 1.970 (3.17), 1.982 (1.57), 1.994 (2.62), 2.006 (1.90), 2.038 (1.64), 2.057 (2.56), 2.065 (1.57), 2.081 (2.75), 2.097 (1.35), 2.121 (1.21 ), 2.144 (1.04), 2.156 (1.46), 2.168 (1.72), 2.181 (1.71 ), 2.190 (1.70), 2.229 (0.47), 3.640 (0.79), 3.656 (1.32), 3.673 (2.34), 3.689 (2.73), 3.706 (1.75), 3.718 (1.78), 3.734 (2.68), 3.751 (2.16), 3.768 (1.32), 3.783 (0.71 ), 7.466 (2.15), 7.484 (4.72), 7.503 (2.97), 7.537 (14.63), 7.542 (16.00), 7.551 (1 1.33), 7.553 (1 1.19), 7.574 (0.99), 7.623
(3.21 ) , 7.679 (8.09), 7.684 (9.17), 7.692 (9.95), 7.703 (6.72), 7.712 (5.60), 7.732 (9.29), 7.753 (1 1.58), 7.772 (3.08), 7.951 (3.64), 7.956 (3.38), 7.973 (6.89), 7.978 (6.87), 8.018 (1 1.35), 8.040 (5.77), 9.055 (2.33), 9.069 (4.61 ), 9.084 (2.26).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.06 (br. s, 1 H), 9.07 (t, 1 H), 8.03 (d, 1 H), 7.97 (dd, 1 H), 7.81-7.43 (m, 10H), 3.87-3.56 (m, 2H), 3.4-3.3 (1 H, verdeckt), 2.26-1.82 (m, 4H).
Beispiel 216
(-)-5-{[(6-Brom-3-chlor-2-phenylchinolin-4-yl)carbonyl]amino}-4-[2-(trffl
säure (Enantiomer 1)
Zu einer Lösung aus (-)-ie/f-Butyl-5-{[(6-brom-3-chlor-2-phenylchinolin-4-yl)carbonyl]amino}-4-[2- (trifluormethyl)phenyl]pentanoat (85 mg, 128 μηηοΙ, Beispiel 281 A) in Dichlormethan (1.1 ml) wurde TFA (220 μΙ, 2.8 mmol) gegeben, und das Gemisch wurde über Nacht bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 17) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 62 mg (100% Reinheit, 80% d. Th.) der Titelverbindung erhalten.
[a]D 20 = -15.0°, 589 nm, c = 0.22 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.13 min; MS (ESIpos): m/z = 605/607 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.236 (1.00), 1.917 (1.50), 1.943 (3.67), 1.957 (2.00), 1.974 (3.20), 1.987 (1.36), 1.998 (2.60), 2.010 (1.91 ), 2.041 (1.57), 2.060 (2.55), 2.068 (1.50), 2.084 (2.74), 2.100 (1.29), 2.123 (1.22), 2.146 (0.98), 2.158 (1.36), 2.170 (1 .63), 2.182 (1.54), 2.192 (1.54), 2.231 (0.45), 2.328 (0.46), 2.670 (0.50), 3.641 (0.73), 3.657 (1.19), 3.674 (2.22), 3.690 (2.57), 3.706 (1.64), 3.718 (1.67), 3.733 (2.53), 3.750 (2.02), 3.767 (1.19), 3.783 (0.62), 7.467 (2.09), 7.485 (4.66), 7.504 (2.87), 7.536 (14.83), 7.542 (16.00), 7.550 (10.71 ), 7.553 (1 1.29), 7.562 (3.32), 7.577 (0.86), 7.623 (3.10), 7.669 (1.40), 7.678 (8.29), 7.683 (9.37), 7.692 (9.37), 7.702 (6.59), 7.713 (5.25), 7.733 (9.14), 7.754 (1 1.35), 7.773 (2.88), 7.951 (3.80), 7.956 (3.60), 7.973 (7.06), 7.978 (7.33), 8.018 (12.32), 8.040 (6.20), 9.049 (2.26), 9.064 (4.63), 9.079
(2.22) , 12.023 (2.74).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.02 (br. s, 1 H), 9.06 (t, 1 H), 8.03 (d, 1 H), 7.97 (dd, 1 H), 7.81-7.44 (m, 10H), 3.82-3.60 (m, 2H), 3.4-3.3 (m, 1 H, teilweise verdeckt), 2.26-1.85 (m, 4H).
Beispiel 217
(+)-5-{[(6-Brom-3-chlor-2-phenylchinolin-4-yl)cato^
säure (Enantiomer 2)
Zu einer Lösung aus (+)-ie/f-Butyl-5-{[(6-brom-3-chlor-2-phenylchinolin-4-yl)carbonyl]amino}-4- [2-(trifluormethyl)phenyl]pentanoat (85 mg, 128 μmol, Beispiel 282A) in Dichlormethan (1.1 ml) wurde TFA (220 μΙ, 2.8 mmol) gegeben, und das Gemisch wurde über Nacht bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 17) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wur- den 62 mg (100% Reinheit, 80% d. Th.) der Titelverbindung erhalten.
[a]D 20 = +14.3°, 589 nm, c = 0.43 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.13 min; MS (ESIpos): m/z = 605/607 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.235 (0.71 ), 1.917 (1.81 ), 1.941 (3.98), 1.955 (2.36), 1.972 (3.30), 1.984 (1.60), 1.995 (2.69), 2.007 (1.95), 2.039 (1.80), 2.058 (2.66), 2.082 (2.81 ), 2.098 (1.37), 2.122 (1.27), 2.145 (1.18), 2.158 (1.55), 2.169 (1.82), 2.181 (1.75), 2.191 (1.71 ), 2.328 (0.42), 3.640 (0.93), 3.655 (1.46), 3.673 (2.46), 3.689 (2.81 ), 3.706 (1.88), 3.718 (1.89), 3.734 (2.72), 3.750 (2.15), 3.767 (1.29), 7.466 (2.34), 7.485 (4.91 ), 7.503 (3.34), 7.536 (15.16), 7.542 (16.00), 7.550 (1 1.28), 7.553 (1 1.15), 7.562 (3.43), 7.575 (1.00), 7.623 (3.41 ), 7.678 (8.94), 7.684 (9.79), 7.692 (10.30), 7.702 (7.12), 7.712 (5.76), 7.732 (9.48), 7.753 (1 1.47), 7.772 (2.96), 7.950 (3.82), 7.956 (3.60), 7.973 (6.94), 7.978 (6.88), 8.018 (1 1.31 ), 8.040 (5.66), 9.052 (2.48), 9.067 (4.61 ), 9.081 (2.21 ), 12.038 (0.76).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.04 (br. s, 1 H), 9.07 (t, 1 H), 8.03 (d, 1 H), 7.97 (dd, 1 H), 7.80-7.43 (m, 10H), 3.81-3.60 (m, 2H), 3.4-3.3 (m, 1 H, teilweise verdeckt), 2.26-1.86 (m, 4H).
Beispiel 218
(+/-)-4-(2-Chlorphenyl)-5-{[(6-ethinyl-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}pentansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-4-(2-chlorphenyl)-5-{[(6-ethinyl-3-methyl-2-phenylchinolin-4- yl)carbonyl]amino}pentanoat (130 mg, 235 μηηοΙ, Beispiel 283A) in Dichlormethan (2.0 ml) wurde TFA (400 μΙ, 5.2 mmol) gegeben, und das Gemisch wurde 3.5 h bei RT stehen gelassen. An- schließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 17) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 69 mg (100% Reinheit, 59% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.88 min; MS (ESIpos): m/z = 497 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.801 (0.56), 1.822 (1.23), 1.844 (1.55), 1.867 (0.84), 2.028 (0.84), 2.048 (3.24), 2.082 (3.52), 2.098 (3.73), 2.126 (4.59), 2.144 (3.85), 2.168 (1.41 ), 3.61 1 (1.29), 3.670 (0.59), 3.683 (0.89), 3.703 (1.37), 3.716 (1.78), 3.729 (1.06), 3.767 (1.15), 3.782 (0.98), 4.414 (6.20), 7.239 (1.05), 7.258 (2.20), 7.277 (1.56), 7.353 (1.29), 7.371 (2.26), 7.389 (1.23), 7.429 (3.77), 7.449 (3.09), 7.497 (6.68), 7.507 (4.98), 7.519 (16.00), 7.706 (2.76), 7.728 (3.22), 7.956 (4.41 ), 7.977 (3.77), 8.822 (1.51 ), 8.837 (2.81 ), 8.850 (1.42).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.06 (br. s, 1 H), 8.84 (t, 1 H), 7.97 (d, 1 H), 7.72 (d, 1 H), 7.7-7.4 (br. m, 1 H), 7.56-7.47 (m, 6H), 7.44 (d, 1 H), 7.37 (t, 1 H), 7.26 (t, 1 H), 4.41 (s, 1 H), 3.90- 3.47 (m, 3H), 2.31-1.99 (m, 6H), 1.94-1.61 (m, 1 H).
Beispiel 219
(-)-4-(2-Chlorphenyl)-5-{[(6-ethinyl-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}pentansäure (Enantiomer 1)
Zu einer Lösung aus (-)-ierf-Butyl-4-(2-chlorphenyl)-5-{[(6-ethinyl-3-methyl-2-phenylchinolin-4- yl)carbonyl]amino}pentanoat (185 mg, 334 μηηοΙ, Beispiel 284A) in Dichlormethan (2.8 ml) wurde TFA (570 μΙ, 7.4 mmol) gegeben, und das Gemisch wurde über Nacht bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 17) gereinigt. Die
vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 126 mg (100% Reinheit, 76% d. Th.) der Titelverbindung erhalten.
[a]D 20 = -20.3°, 589 nm, c = 0.41 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.88 min; MS (ESIpos): m/z = 497 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.802 (0.53), 1.823 (1.17), 1.845 (1.47), 1.867 (0.80), 2.050 (3.23), 2.085 (3.29), 2.100 (3.72), 2.128 (4.42), 2.147 (3.44), 2.171 (1.30), 3.612 (1.21 ), 3.671 (0.55), 3.684 (0.83), 3.703 (1.29), 3.717 (1.70), 3.730 (1.00), 3.766 (1.09), 4.414 (7.46), 7.238 (0.99), 7.258 (2.14), 7.277 (1.51 ), 7.353 (1.23), 7.371 (2.17), 7.389 (1.18), 7.429 (3.88), 7.449 (3.19), 7.498 (6.54), 7.507 (4.77), 7.519 (16.00), 7.706 (3.04), 7.728 (3.50), 7.956 (4.48), 7.978 (3.78), 8.821 (1.46), 8.835 (2.75), 8.849 (1.39), 12.049 (0.97).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.05 (br. s, 1 H), 8.84 (t, 1 H), 7.97 (d, 1 H), 7.72 (d, 1 H), 7.7-7.4 (br. m, 1 H), 7.56-7.47 (m, 6H), 7.44 (d, 1 H), 7.37 (t, 1 H), 7.26 (t, 1 H), 4.41 (s, 1 H), 3.87- 3.48 (m, 3H), 2.27-1.95 (m, 6H), 1.92-1.70 (m, 1 H).
Beispiel 220
(+)-4-(2-Chlorphenyl)-5-{[(6-ethinyl-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}pentansäure (Enantiomer 2)
Zu einer Lösung aus (+)-ierf-Butyl-4-(2-chlorphenyl)-5-{[(6-ethinyl-3-methyl-2-phenylchinolin-4- yl)carbonyl]amino}pentanoat (185 mg, 334 μηηοΙ, Beispiel 285A) in Dichlormethan (2.8 ml) wurde TFA (570 μΙ, 7.4 mmol) gegeben, und das Gemisch wurde über Nacht bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 17) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 152 mg (100% Reinheit, 91 % d. Th.) der Titelverbindung erhalten.
[a]D 20 = +20.2°, 589 nm, c = 0.32 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.88 min; MS (ESIpos): m/z = 497 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.802 (0.55), 1.823 (1.21 ), 1.845 (1.53), 1.868 (0.84), 2.030 (0.81 ), 2.038 (0.93), 2.051 (3.31 ), 2.086 (3.46), 2.101 (3.94), 2.128 (4.59), 2.148 (3.59), 2.171 (1.36), 3.613 (1.28), 3.670 (0.59), 3.684 (0.90), 3.703 (1.37), 3.717 (1.79), 3.730 (1.07), 3.750 (0.88), 3.766 (1.13), 3.782 (0.97), 4.414 (6.81 ), 7.239 (1.01 ), 7.258 (2.19), 7.277 (1.54), 7.353 (1.26), 7.372 (2.24), 7.390 (1.22), 7.429 (3.97), 7.449 (3.25), 7.498 (6.77), 7.507 (4.77), 7.519 (16.00), 7.706 (2.80), 7.728 (3.25), 7.956 (4.78), 7.978 (4.08), 8.820 (1.55), 8.835 (2.96), 8.849 (1.51 ).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.04 (br. s, 1 H), 8.83 (t, 1 H), 7.97 (d, 1 H), 7.72 (d, 1 H), 7.7-7.4 (br. m, 1 H), 7.57-7.47 (m, 6H), 7.44 (d, 1 H), 7.37 (t, 1 H), 7.26 (t, 1 H), 4.41 (s, 1 H), 3.88- 3.53 (m, 3H), 2.25-1.96 (m, 6H), 1.93-1.71 (m, 1 H).
Beispiel 221
(+/-)-5-{[(6-Brom-3-chlor-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-cr^
pentansäure (Racemat)
Zu einer Lösung aus (+/-)-ie/f-Butyl-5-{[(6-brom-3-chlor-2-phenylchinolin-4-yl)carbonyl]amino}-4- (2-chlor-3,6-difluorphenyl)pentanoat (65 mg, 98 μηηοΙ, Beispiel 286A) in Dichlormethan (830 μΙ) wurde TFA (170 μΙ, 2.2 mmol) gegeben, und das Gemisch wurde 18 h bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 17) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 34 mg (100% Reinheit, 56% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.08 min; MS (ESIpos): m/z = 607/609/61 1 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.981 (1.03), 2.074 (1.54), 2.093 (1.50), 2.109 (3.76), 2.146 (8.52), 2.174 (2.16), 2.189 (1.98), 2.212 (0.70), 2.695 (0.74), 3.755 (2.13), 3.774 (2.01 ), 3.819 (2.95), 7.268 (1.40), 7.279 (1.57), 7.291 (2.84), 7.302 (2.94), 7.317 (2.27), 7.328 (2.13), 7.382 (1.83), 7.393 (2.1 1 ), 7.404 (2.97), 7.415 (2.88), 7.425 (1.53), 7.437 (1.31 ), 7.531 (14.68), 7.537 (16.00), 7.547 (12.47), 7.569 (0.76), 7.657 (8.52), 7.663 (9.38), 7.672 (8.91 ), 7.681 (6.87), 7.694 (3.13), 7.956 (3.51 ), 7.961 (3.33), 7.978 (7.36), 7.983 (7.60), 8.015 (12.42), 8.037 (5.55), 9.088 (2.21 ), 9.102 (4.20), 9.1 15 (2.13), 12.096 (8.00).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.10 (s, 1 H), 9.10 (t, 1 H), 8.02 (d, 1 H), 7.97 (dd, 1 H), 7.73-7.63 (m, 3H), 7.57-7.51 (m, 3H), 7.41 (td, 1 H), 7.30 (td, 1 H), 3.95-3.67 (m, 3H), 2.27-2.05 (m, 3H), 2.04-1.89 (m, 1 H).
Beispiel 222
(-)-5-{[(6-Brom-3-chlor-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-ch
säure (Enantiomer 1)
Zu einer Lösung aus (-)-ie/f-Butyl-5-{[(6-brom-3-chlor-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2- chlor-3,6-difluorphenyl)pentanoat (45 mg, 68 μηηοΙ, Beispiel 287A) in Dichlormethan (520 μΙ) wurde TFA (1 10 μΙ, 1.5 mmol) gegeben, und das Gemisch wurde 22 h bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 17) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 34 mg (100% Reinheit, 82% d. Th.) der Titelverbindung erhalten.
[a]D 20 = -33.3°, 589 nm, c = 0.38 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.08 min; MS (ESIpos): m/z = 607/609/61 1 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.235 (0.82), 1.980 (1.02), 2.093 (1.51 ), 2.109 (3.71 ), 2.146 (8.29), 2.189 (1.92), 2.212 (0.68), 3.753 (2.12), 3.775 (2.01 ), 3.819 (2.95), 7.268 (1.36), 7.279 (1.56), 7.292 (2.78), 7.303 (2.89), 7.317 (2.23), 7.329 (2.09), 7.382 (1.79), 7.393 (2.05), 7.404 (2.89), 7.415 (2.80), 7.426 (1.49), 7.437 (1.25), 7.522 (1.82), 7.531 (14.85), 7.538 (16.00), 7.546 (1 1.67), 7.548 (12.40), 7.569 (0.72), 7.649 (1.59), 7.658 (8.50), 7.663 (9.30), 7.672 (8.78), 7.681 (6.87), 7.694 (3.02), 7.956 (3.50), 7.961 (3.35), 7.978 (7.36), 7.984 (7.66), 8.015 (12.07), 8.037 (5.42), 9.088 (2.19), 9.102 (4.15), 9.1 16 (2.11 ), 12.097 (2.42).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.10 (s, 1 H), 9.10 (t, 1 H), 8.02 (d, 1 H), 7.97 (dd, 1 H), 7.74-7.63 (m, 3H), 7.58-7.51 (m, 3H), 7.41 (td, 1 H), 7.30 (td, 1 H), 3.94-3.67 (m, 3H), 2.26-2.06 (m, 3H), 1.98 (br. s, 1 H).
Beispiel 223
(+)-5-{[(6-Brom-3-chlor-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2-chlor-3,6-difluorphenyl)- pentansäure (Enantiomer 2)
Zu einer Lösung aus (+)-ie/f-Butyl-5-{[(6-brom-3-chlor-2-phenylchinolin-4-yl)carbonyl]amino}-4- (2-chlor-3,6-difluorphenyl)pentanoat (45 mg, 68 μηηοΙ, Beispiel 288A) in Dichlormethan (520 μΙ) wurde TFA (1 10 μΙ, 1.5 mmol) gegeben, und das Gemisch wurde 22 h bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 17) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 29 mg (100% Reinheit, 69% d. Th.) der Titelverbindung erhalten.
[a]D 20 = +35.3°, 589 nm, c = 0.36 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.08 min; MS (ESIpos): m/z = 607/609/61 1 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (0.62), 1.235 (0.56), 1.977 (0.97), 2.108 (3.49), 2.142 (8.02), 2.186 (1.89), 2.209 (0.65), 3.750 (2.03), 3.774 (1.91 ), 3.817 (2.76), 7.267 (1.37), 7.278 (1.54), 7.291 (2.80), 7.302 (2.88), 7.317 (2.23), 7.328 (2.12), 7.381 (1.76), 7.393 (2.01 ), 7.403 (2.88), 7.414 (2.78), 7.425 (1.49), 7.436 (1.23), 7.522 (1.67), 7.531 (15.31 ), 7.537 (16.00), 7.547 (12.27), 7.555 (3.68), 7.569 (0.74), 7.648 (1.53), 7.657 (8.76), 7.662 (9.30), 7.672 (8.81 ), 7.681 (6.84), 7.693 (3.00), 7.956 (3.75), 7.961 (3.45), 7.978 (7.82), 7.983 (7.97), 8.015 (12.92), 8.037 (5.78), 9.088 (2.12), 9.102 (4.02), 9.1 16 (2.04), 12.106 (0.55).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.1 1 (br. s, 1 H), 9.10 (t, 1 H), 8.02 (d, 1 H), 7.97 (dd, 1 H), 7.74-7.63 (m, 3H), 7.58-7.50 (m, 3H), 7.41 (td, 1 H), 7.30 (td, 1 H), 3.94-3.67 (m, 3H), 2.25- 2.06 (m, 3H), 1.98 (br. s, 1 H).
Beispiel 224
(+/-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(pyridin
(Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 4-(pyridin-2-yl)pentanoat (100 mg, 174 μηηοΙ, Beispiel 289A, nicht reinheitskorrigiert) in Dichlormethan (750 μΙ) wurde TFA (270 μΙ, 3.5 mmol) gegeben, und das Gemisch wurde 7 h bei RT gerührt. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Säule Chromatorex C18, 10 m, 125 x 30 mm; Gradient 20% -> 60% Acetonitril in 0.1 % wässriger Ammoniaklösung) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 46 mg (92% Reinheit, 49% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 5): Rt = 0.99 min; MS (ESIpos): m/z = 518/520 [M+H]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.107 (12.82), 1.737 (0.48), 1.882 (0.41 ), 1.905 (0.87), 1.914 (0.67), 1.924 (1.29), 1.938 (0.95), 1.945 (0.81 ), 1.967 (0.78), 1.982 (0.86), 1.988 (1.35), 1.995 (1.12), 2.000 (1.1 1 ), 2.01 1 (1.20), 2.021 (2.54), 2.047 (2.18), 2.059 (1.78), 2.067 (1.57),
2.079 (2.19), 2.102 (2.39), 2.108 (2.79), 2.125 (4.18), 2.147 (2.08), 2.518 (2.49), 2.523 (1.57), 2.539 (1.37), 3.202 (0.98), 3.619 (0.83), 3.638 (0.87), 3.652 (1.1 1 ), 3.794 (0.70), 7.247 (1.05), 7.263 (1.42), 7.276 (1.14), 7.312 (1.73), 7.331 (1.84), 7.472 (0.45), 7.475 (0.48), 7.481 (0.73), 7.485 (1.28), 7.487 (1.44), 7.498 (4.79), 7.506 (4.67), 7.516 (16.00), 7.518 (15.39), 7.523 (7.66), 7.528 (5.83), 7.532 (5.46), 7.539 (1.18), 7.737 (1.28), 7.752 (1.95), 7.772 (1.02), 7.829 (2.64), 7.834 (2.35), 7.851 (3.81 ), 7.856 (3.77), 7.922 (7.82), 7.944 (4.82), 8.579 (2.37), 8.589 (2.35), 8.796 (1.37), 8.81 1 (2.19), 8.825 (1.33), 12.041 (1.59).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.04 (br. s, 1 H), 8.81 (t, 1 H), 8.58 (d, 1 H), 7.93 (d, 1 H), 7.84 (dd, 1 H), 7.75 (t, 1 H), 7.67 (br. s, 1 H), 7.55-7.45 (m, 5H), 7.32 (d, 1 H), 7.26 (t, 1 H), 3.88- 3.73 (m, 1 H), 3.71-3.55 (m, 1 H), 3.27-3.13 (m, 1 H), 2.23-1.86 (m, 7H).
Beispiel 225
(+/-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-[2-fluor-6-(trffl
phenyl]pentansäure (Racemat)
(+/-)-fe/f-Butyl-5-{[(6-brom-3-methyl-2-ph^
methyl)phenyl]pentanoat (80 mg, 93% Reinheit, 1 13 μηηοΙ, Beispiel 290A) wurde in Dichlorme- than (1.5 ml) gelöst. Bei RT wurde TFA (850 μΙ, 1 1.28 mmol) zugegeben und das Gemisch 1 h bei RT gerührt. Anschließend wurden die flüchtigen Komponenten am Rotationsverdampfer entfernt. Der Rückstand wurde dreimal mit etwas Toluol versetzt und erneut am Rotationsverdamp- fer eingeengt. Der Rückstand wurde in wenig DMSO gelöst und mittels präparativer HPLC (Methode 28) gereinigt. Die geeigneten Fraktionen wurden vereinigt und im Vakuum eingeengt. Der Rückstand wurde im Vakuum getrocknet. Es wurden 63 mg (100% Reinheit, 93% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.03 min; MS (ESIpos): m/z = 603/605 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.1 1 (br. s, 1 H), 8.96 (t, 1 H), 7.95 (d, 1 H), 7.85 (dd, 1 H), 7.75-7.46 (m, 9H), 3.95 (br. s, 1 H), 3.72 (br. s, 1 H), 3.31 (1 H, verdeckt, tentativ), 2.25-1.98 (m, 7H).
Beispiel 226
(+)-5-{[(6-Brom-3-methyl-2-phenylchinolin^
phenyl]pentansäure (Enantiomer 1)
ie f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-[2-fluor-6-(trffl
phenyljpentanoat (440 mg, 667 μηηοΙ, Beispiel 291 A, Enantiomer 1) wurde in Dichlormethan (8.9 ml) gelöst. Bei RT wurde TFA (5 ml, 66.7 mmol) zugegeben und das Gemisch 1 h bei RT gerührt. Anschließend wurden die flüchtigen Komponenten am Rotationsverdampfer entfernt. Der Rückstand wurde in wenig DMSO gelöst und mittels präparativer HPLC (Methode 29) gereinigt. Die geeigneten Fraktionen wurden vereinigt und in Vakuum eingeengt. Der Rückstand wurde im Vakuum getrocknet. Es wurden 356 mg (98% Reinheit, 87% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.01 min; MS (ESIpos): m/z = 603/605 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.06 (br. s, 1 H), 8.95 (t, 1 H), 7.95 (d, 1 H), 7.85 (dd, 1 H), 7.78-7.44 (m, 9H), 3.96 (br. s, 1 H), 3.73 (br. s, 1 H), 3.32 (1 H, verdeckt, tentative), 2.24-2.00 (m, 7H).
[a]D 20 = +45.6°, 589 nm, c = 0.415 g/100 ml, Methanol Beispiel 227
(-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-[2-fluor-6-(trifluor- methyl)phenyl]pentansäure (Enantiomer 2)
tert-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-[2-fluor-6-(trifluormethyl)- phenyl]pentanoat (412 mg, 625 μηηοΙ, Beispiel 292A, Enantiomer 2) wurde in Dichlormethan (8.3 ml) gelöst. Bei RT wurde TFA (4.6 ml, 62.5 mmol) zugegeben und das Gemisch 1 h bei RT gerührt. Anschließend wurden die flüchtigen Komponenten am Rotationsverdampfer entfernt. Der Rückstand wurde in wenig DMSO gelöst und über präparativer HPLC (Methode 29) gereinigt. Die geeigneten Fraktionen wurden vereinigt und in Vakuum eingeengt. Der Rückstand wurde im Vakuum getrocknet. Es wurden 344 mg (98% Reinheit, 89% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.01 min; MS (ESIpos): m/z = 603/605 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.06 (br. s, 1 H), 8.95 (t, 1 H), 7.95 (d, 1 H), 7.85 (dd, 1 H), 7.80-7.44 (m, 9H), 3.96 (br. s, 1 H), 3.73 (br. s, 1 H), 3.33 (1 H, teilweise verdeckt, tentative), 2.24-2.00 (m, 7H).
[a]D 20 = -45.6°, 589 nm, c = 0.38 g/100 ml, Methanol
Beispiel 228
(+/-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)c^^
pentansäure (Racemat)
(+/-)-ie f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amin
phenyl)pentanoat (25 mg, 38.7 μηηοΙ, Beispiel 293A) wurde in Dichlormethan (520 μΙ) gelöst. Bei RT wurde TFA (290 μΙ, 3.8 mmol) zugegeben und das Gemisch über Nacht bei RT gerührt. Anschließend wurden die flüchtigen Komponenten am Rotationsverdampfer entfernt. Der Rückstand wurde mehrfach mit etwas Toluol versetzt und erneut am Rotationsverdampfer eingeengt. Der Rückstand wurde in wenig DMSO gelöst und mittels präparativer HPLC (Methode 28) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 17 mg (100% Reinheit, 74% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.04 min; MS (ESIpos): m/z = 589/591 [M+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 12.13 (br. s, 1 H), 8.98 (t, 1 H), 7.95 (d, 1 H), 7.86 (dd, 1 H), 7.8-7.6 (m, 2H), 7.58-7.46 (m, 5H), 3.87-3.77 (m, 2H), 3.61-3.51 (m, 1 H), 2.27-2.04 (m, 6H), 2.00-1.90 (m, 1 H).
Beispiel 229
(-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2,3,5,6-tetrafluorphenyl)- pentansäure (Enantiomer 1)
ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2,3,5,6-tetrafluor- phenyl)pentanoat (265 mg, 410 μηηοΙ, Beispiel 294A, Enantiomer 1) wurde in Dichlormethan (5.5 ml) gelöst. Bei RT wurde TFA (3.1 ml, 41.0 mmol) zugegeben und das Gemisch über Nacht bei RT gerührt. Anschließend wurden die flüchtigen Komponenten am Rotationsverdampfer entfernt. Der Rückstand wurde mehrfach mit etwas Toluol versetzt und erneut am Rotationsver- dampfer eingeengt. Der Rückstand wurde in wenig DMSO gelöst und mittels präparativer HPLC (Methode 28) gereinigt. Die geeigneten Fraktionen wurden vereinigt und im Vakuum eingeengt.
Der Rückstand wurde im Vakuum getrocknet. Es wurden 237 mg (98% Reinheit, 96% d. Th.) der Titelverbindung erhalten.
[a]D 20 = -50.9°, 589 nm, c = 0.45 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 1.99 min; MS (ESIpos): m/z = 589/591 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.12), 0.008 (1.97), 1.922 (0.60), 1.956 (1.02), 1.981 (0.92), 1.997 (0.53), 2.074 (1.06), 2.088 (1.41 ), 2.107 (1.34), 2.128 (1.19), 2.158 (6.34), 2.202 (3.94), 2.219 (5.66), 2.238 (2.23), 2.328 (0.54), 2.366 (0.44), 2.669 (0.47), 2.710 (0.41 ), 3.167 (0.95), 3.558 (1.10), 3.794 (2.43), 3.810 (3.34), 7.495 (1.52), 7.506 (4.54), 7.514 (4.40), 7.525 (16.00), 7.536 (6.45), 7.539 (5.76), 7.800 (1.00), 7.848 (2.66), 7.853 (2.35), 7.870 (3.85), 7.875 (3.72), 7.940 (7.13), 7.963 (4.44), 8.155 (0.67), 8.969 (1.31 ), 8.984 (2.59), 8.998 (1.30).
Beispiel 230
(+)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2,3,5,6-tetrafluorphenyl)- pentansäure (Enantiomer 2)
ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2,3,5,6-tetrafluor- phenyl)pentanoat (274 mg, 99% Reinheit, 420 μηηοΙ, Beispiel 295A, Enantiomer 2) wurde in Dichlormethan (5.6 ml) gelöst. Bei RT wurde TFA (3.2 ml, 42.0 mmol) zugegeben und das Gemisch über Nacht bei RT gerührt Anschließend wurden die flüchtigen Komponenten am Rotationsverdampfer entfernt. Der Rückstand wurde mehrfach mit etwas Toluol versetzt und erneut am Rotationsverdampfer eingeengt. Der Rückstand wurde in wenig DMSO gelöst und mittels präparativer HPLC (Methode 28) gereinigt. Die geeigneten Fraktionen wurden vereinigt und im Vakuum eingeengt. Der Rückstand wurde im Vakuum getrocknet. Es wurden 235 mg (99% Reinheit, 94% d. Th.) der Titelverbindung erhalten.
[a]D 20 = +47.2°, 589 nm, c = 0.445 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.00 min; MS (ESIpos): m/z = 589/591 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.64), 0.008 (1.47), 1.923 (0.61 ), 1.957 (1.04), 1.981 (0.93), 1.998 (0.52), 2.056 (0.44), 2.075 (1.06), 2.089 (1.38), 2.107 (1.35), 2.126 (1.21 ), 2.159 (6.40), 2.202 (3.92), 2.219 (5.62), 2.238 (2.22), 2.329 (0.43), 3.559 (1.10), 3.794 (2.44), 3.809 (3.34), 7.495 (1.49), 7.506 (4.49), 7.514 (4.34), 7.525 (16.00), 7.536 (6.42), 7.539 (5.68), 7.799 (1.00), 7.848 (2.55), 7.853 (2.31 ), 7.870 (3.72), 7.875 (3.60), 7.941 (6.90), 7.963 (4.29), 8.969 (1.33), 8.984 (2.61 ), 8.998 (1.28), 12.153 (0.46).
Beispiel 231
(+/-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2,3,5-trifluorphenyl)- pentansäure (Racemat)
(+/-)-ie f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]am
phenyl)pentanoat (25 mg, 39.8 μmol, Beispiel 296A) wurde in Dichlormethan (530 μΙ) gelöst. Bei RT wurde TFA (40 μΙ, 3.1 mmol) zugegeben und das Gemisch 1 h bei RT gerührt. Da die Um- Setzung nicht vollständig war (HPLC Kontrolle), wurden zusätzliche Portionen Trifluoressigsäure (insgesamt 230 μΙ) hinzugegeben. Es wurde weiter gerührt, bis kein Ausgangsmaterial per HPLC nachweisbar war (4 h). Anschließend wurden die flüchtigen Komponenten am Rotationsverdampfer entfernt. Der Rückstand wurde mehrfach mit etwas Toluol versetzt und erneut am Rotationsverdampfer eingeengt. Der Rückstand wurde in wenig DMSO gelöst und mittels präparati- ver HPLC (Methode 28) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 20 mg (100% Reinheit, 88% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.02 min; MS (ESIpos): m/z = 571/573 [M+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 12.10 (br. s, 1 H), 8.89 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.81-7.56 (br. s, 1 H), 7.56-7.46 (m, 5H), 7.46-7.35 (m, 1 H), 7.27-7.20 (m, 1 H), 3.84-3.66 (m, 2H), 3.50-3.37 (m, 1 H), 2.24-1.98 (m, 6H), 1.88-1.77 (m, 1 H).
Beispiel 232
5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2,3,5-trifluorphenyl)pentansäure (Enantiomer 1)
ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2,3,5-trifluorphenyl)- pentanoat (38 mg, 60.2 μηηοΙ, Beispiel 297A, Enantiomer 1) wurde in Dichlormethan (800 μΙ) gelöst. Bei RT wurde TFA (450 μΙ) hinzugegeben und das Gemisch über Nacht bei RT gerührt. Anschließend wurden die flüchtigen Komponenten am Rotationsverdampfer entfernt. Der Rückstand wurde mehrfach mit etwas Toluol versetzt und erneut am Rotationsverdampfer eingeengt. Der Rückstand wurde in wenig DMSO gelöst und mittels präparativer HPLC (Methode 28) gerei-
nigt. Die vereinigten Zielfraktionen wurden im Vakuum eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 29 mg (100% Reinheit, 84% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 1.04 min; MS (ESIpos): m/z = 571/573 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (2.16), 1.798 (0.71 ), 1.834 (1.03), 1.849 (0.83), 1.872 (0.53), 2.018 (1.07), 2.031 (1.34), 2.051 (1.41 ), 2.073 (9.44), 2.082 (1.24), 2.124 (4.71 ), 2.145 (7.02), 2.327 (0.68), 2.366 (0.51 ), 2.670 (0.61 ), 2.709 (0.50), 3.428 (1.17), 3.682 (0.83), 3.703 (1.26), 3.716 (1.82), 3.729 (1.15), 3.763 (0.91 ), 7.223 (1.35), 7.41 1 (1.03), 7.494 (1.56), 7.504 (4.57), 7.512 (4.41 ), 7.523 (16.00), 7.534 (6.76), 7.837 (2.19), 7.842 (2.04), 7.859 (3.36), 7.864 (3.29), 7.933 (6.54), 7.956 (4.15), 8.875 (1.32), 8.890 (2.48), 8.903 (1.37), 12.092 (0.68). Beispiel 233
5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2,3,5-trifluorphenyl)pentansäure (Enantiomer 2)
ierf-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2,3,5-trifluorphenyl)penta- noat (40 mg, 62.8 μηηοΙ, Beispiel 298A, Enantiomer 2) wurde in Dichlormethan (840 μΙ) gelöst. Bei RT wurde Trifluoressigsäure (470 μΙ) hinzugegeben und das Gemisch über Nacht bei RT gerührt. Anschließend wurden die flüchtigen Komponenten am Rotationsverdampfer entfernt. Der Rückstand wurde mehrfach mit etwas Toluol versetzt und erneut am Rotationsverdampfer eingeengt. Der Rückstand wurde in wenig DMSO gelöst und über präparativer HPLC (Methode 28) gereinigt. Die vereinigten Zielfraktionen wurden im Vakuum eingeengt, und der Rückstand wurde im Vaku- um getrocknet. Es wurden 25 mg (100% Reinheit, 70% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 1.97 min; MS (ESIpos): m/z = 571/573 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.149 (1.73), -0.008 (16.00), 0.008 (15.95), 0.146 (1.73), 1.825 (0.98), 2.022 (1.21 ), 2.041 (1.17), 2.060 (1.31 ), 2.1 17 (3.50), 2.140 (3.50), 2.327 (1.77), 2.366 (1.82), 2.670 (1.96), 2.710 (2.01 ), 3.41 1 (1.35), 3.713 (1.63), 7.225 (1.35), 7.395 (1.07), 7.503 (4.34), 7.511 (4.10), 7.523 (14.23), 7.836 (2.19), 7.841 (2.01 ), 7.859 (3.17), 7.864 (3.08), 7.932 (6.30), 7.955 (4.10), 8.908 (2.10).
Beispiel 234
(+/-)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2,3,6-trichlorphenyl)pentan- säure (Racemat)
(+/-)-ie f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]am
nyl)pentanoat (80 mg, 1 18 μmol, Beispiel 299A) wurde in Dichlormethan (1.6 ml) gelöst. Bei RT wurde TFA (880 μΙ, 1 1.8 mmol ) zugegeben, und das Gemisch wurde 2 h bei RT gerührt. Die flüchtigen Komponenten wurden am Rotationsverdampfer entfernt, und der Rückstand wurde im Vakuum getrocknet. Anschließend wurde das Rohprodukt in wenig DMSO gelöst und mittels präparativer HPLC (Methode 29) gereinigt. Die vereinigten Zielfraktionen wurden im Vakuum eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 66 mg (100% Reinheit, 90% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.14 min; MS (ESIpos): m/z = 619/621/623 [M+H]+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 12.11 (br. s, 1 H), 8.99-8.87 (m, 1 H), 7.95 (dd, 1 H), 7.89-7.83 (m, 1 H), 7.82-7.64 (m, 1 H), 7.64-7.57 (m, 1 H), 7.56-7.46 (m, 6H), 4.20-4.00 (m, 2H), 3.94-3.79 (m, 1 H), 2.35-2.03 (m, 7H).
Beispiel 235
(+)-5-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2,3,6-trichlorphenyl)- pentansäure (Enantiomer 1)
ie/f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-4-(2,3,6-trichlorphenyl)- pentanoat (227 mg, 95% Reinheit, 319 μηηοΙ, Beispiel 300A, Enantiomer 1) wurde in Dichlormethan (2.7 ml) gelöst. Bei RT wurde TFA (2.4 ml) zugegeben und das Gemisch 2 h bei RT gerührt Anschließend wurden die flüchtigen Komponenten am Rotationsverdampfer entfernt. Der Rückstand wurde zweimal mit etwas Toluol versetzt und erneut am Rotationsverdampfer eingeengt. Der Rückstand wurde in wenig DMSO gelöst und mittels präparativer HPLC (Methode 30) gereinigt. Die vereinigten Zielfraktionen wurden im Vakuum eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 148 mg (96% Reinheit, 73% d. Th.) der Titelverbindung erhalten. [a]D 20 = +34.1 °, 589 nm, c = 0.44 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.1 1 min; MS (ESIpos): m/z = 619/621/623 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 2.073 (8.70), 2.095 (2.02), 2.108 (2.74), 2.128 (3.53), 2.153 (5.13), 2.164 (4.58), 2.180 (9.66), 2.212 (1.42), 2.237 (0.76), 2.283 (1.22), 2.31 1 (1.53), 2.328 (1.16), 2.670 (0.67), 3.832 (0.83), 3.850 (1.05), 3.863 (1.16), 3.876 (1.10), 4.087 (1.55), 4.1 1 1 (1.70), 4.141 (0.81 ), 7.476 (1.72), 7.504 (5.34), 7.509 (4.33), 7.513 (4.34), 7.528 (16.00), 7.551 (5.39), 7.586 (2.1 1 ), 7.593 (2.25), 7.607 (1.58), 7.695 (0.45), 7.741 (0.47), 7.840 (1.67), 7.846 (2.40), 7.863 (2.55), 7.868 (3.83), 7.874 (2.07), 7.935 (4.34), 7.940 (3.94), 7.957 (2.78), 7.963 (2.48), 8.927 (1.77), 8.940 (2.09), 12.107 (1.53).
Beispiel 236
(-)-5-{[(6-Brom-3-methyl-2-phenyl^
pentansäure (Enantiomer 2)
ie f-Butyl-5-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]am
pentanoat (280 mg, 99% Reinheit, 410 pmol, Beispiel 301A, Enantiomer 2) wurde in Dichlorme- than (3.5 ml) gelöst. Bei RT wurde TFA (3.1 ml) zugegeben und das Gemisch 2 h bei RT gerührt Anschließend wurden die flüchtigen Komponenten am Rotationsverdampfer entfernt. Der Rück- stand wurde zweimal mit etwas Toluol versetzt und erneut am Rotationsverdampfer eingeengt. Der Rückstand wurde in wenig DMSO gelöst und mittels präparativer HPLC (Methode 31 ) gereinigt. Die vereinigten Zielfraktionen wurden im Vakuum eingeengt, und der Rückstand wurde im Vakuum getrocknet. Es wurden 214 mg (100% Reinheit, 84% d. Th.) der Titelverbindung erhalten. [a]D 20 = -33.9°, 589 nm, c = 0.19 g/100 ml, Methanol
LC-MS (Methode 2): Rt = 1.1 1 min; MS (ESIpos): m/z = 619/621/623 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.149 (0.76), -0.008 (6.72), 0.008 (7.91 ), 0.146 (0.78), 2.073 (5.46), 2.082 (1.42), 2.095 (2.02), 2.108 (2.66), 2.128 (3.53), 2.153 (5.18), 2.164 (4.61 ), 2.180 (9.99), 2.212 (1.38), 2.237 (0.78), 2.284 (1.19), 2.310 (1.54), 2.327 (1.49), 2.366 (0.71 ), 2.670 (1.10), 2.710 (0.64), 3.832 (0.80), 3.863 (1.17), 4.088 (1.56), 4.1 1 1 (1.77), 4.140 (0.85), 7.476 (1.77), 7.504 (5.32), 7.510 (4.24), 7.513 (4.22), 7.528 (16.00), 7.551 (5.89), 7.593 (2.27), 7.607 (1.58), 7.746 (0.46), 7.840 (1.83), 7.846 (2.52), 7.852 (1.40), 7.863 (2.73), 7.869 (4.03), 7.875 (2.13), 7.935 (4.52), 7.940 (4.06), 7.957 (2.87), 7.963 (2.48), 8.926 (1.83), 8.940 (2.27), 12.1 1 1 (1.05). Beispiel 237
(+/-)-6-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-5-(2-chlorphenyl)hexansäure (Racemat)
Zu einer Lösung aus (+/-)-ierf-Butyl-6-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- 5-(2-chlorphenyl)hexanoat (100 mg, 161 μηηοΙ, Beispiel 302A) in Dichlormethan (1.2 ml) wurde TFA (120 μΙ, 1.6 mmol) gegeben, und das Gemisch wurde drei Tage bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde in Acetonitril gelöst und mittels praparativer HPLC (Methode 15) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 73 mg (100% Reinheit, 80% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.02 min; MS (ESIpos): m/z = 565/567 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1 1.97 (br. s, 1 H), 8.85 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.72 (br. s, 1 H), 7.56-7.42 (m, 7H), 7.37 (t, 1 H), 7.26 (t, 1 H), 3.80-3.35 (m, 3H, teilweise verdeckt), 2.27-2.06 (m, 5H), 1.87-1.72 (m, 1 H), 1.70-1.57 (m, 1 H), 1.53-1.25 (m, 2H).
Beispiel 238
(-)-6-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-5-(2-chlorphenyl)hexansäure (Enantiomer 1)
Zu einer Lösung aus ie/f-Butyl-6-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-5-(2- chlorphenyl)hexanoat (460 mg, 740 μηηοΙ, Beispiel 303A, Enantiomer 1) in Dichlormethan (6.2 ml) wurde TFA (1.3 ml, 16 mmol) gegeben, und das Gemisch wurde 16 h bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 367 mg (100% Reinheit, 88% d. Th.) der Titelverbindung erhalten.
[a]D 20 = -12.4°, 589 nm, c = 0.37 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.03 min; MS (ESIpos): m/z = 565/567 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.326 (0.41 ), 1.339 (0.80), 1.358 (1.09), 1.370 (1.18), 1.384 (1.22), 1.398 (0.94), 1.422 (0.87), 1.438 (1.19), 1.449 (1.18), 1.457 (1.1 1 ), 1.464 (1.30),
1.481 (1.04), 1.497 (0.61 ), 1.594 (0.46), 1.628 (1.23), 1.640 (1.09), 1.652 (1.32), 1.664 (1.00), 1.675 (0.67), 1.687 (0.47), 1.742 (0.66), 1.755 (1.22), 1.769 (1.35), 1.782 (1.33), 1.802 (0.95), 1.815 (0.72), 2.150 (6.24), 2.187 (4.80), 2.205 (8.14), 2.224 (3.74), 2.248 (0.42), 3.170 (9.01 ), 3.548 (1.70), 3.587 (3.08), 3.601 (3.03), 3.965 (0.54), 7.235 (1.26), 7.254 (2.76), 7.273 (1.90), 7.349 (1.67), 7.368 (3.04), 7.386 (1.62), 7.432 (4.96), 7.452 (4.08), 7.486 (5.02), 7.502 (8.76), 7.508 (6.18), 7.521 (16.00), 7.525 (15.01 ), 7.723 (0.62), 7.835 (2.89), 7.840 (2.55), 7.857 (4.35), 7.862 (4.06), 7.932 (8.15), 7.954 (5.1 1 ), 8.834 (1.76), 8.849 (3.52), 8.863 (1.67).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1 1.95 (br. s, 1 H), 8.85 (t, 1 H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.72 (br. s, 1 H), 7.56-7.42 (m, 7H), 7.37 (t, 1 H), 7.26 (t, 1 H), 3.77-3.51 (m, 3H, teilweise verdeckt), 2.25-2.09 (m, 5H), 1.85-1.73 (m, 1 H), 1.71-1.56 (m, 1 H), 1.53-1.25 (m, 2H).
Beispiel 239
(+)-6-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-5-(2-ch
(Enantiomer 2)
Zu einer Lösung aus ie/f-Butyl-6-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-5-(2- chlorphenyl)hexanoat (415 mg, 667 μηηοΙ, Beispiel 304A, Enantiomer 2) in Dichlormethan (5.6 ml) wurde TFA (1 1.1 ml, 15 mmol) gegeben, und das Gemisch wurde 16 h bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde mittels praparativer HPLC (Methode 16) gereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde lyophilisiert. Es wurden 367 mg (100% Reinheit, 88% d. Th.) der Titelverbindung erhalten.
[a]D 20 = +13.6°, 589 nm, c = 0.44 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.03 min; MS (ESIpos): m/z = 565/567 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (1.03), 1.337 (0.89), 1.356 (1.15), 1.369 (1.22), 1.382 (1.24), 1.396 (0.97), 1.421 (0.94), 1.436 (1.25), 1.447 (1.20), 1.455 (1.14), 1.462 (1.28), 1.480 (1.00), 1.495 (0.57), 1.592 (0.56), 1.626 (1.26), 1.640 (1.1 1 ), 1.650 (1.31 ), 1.662 (0.99), 1.687 (0.45), 1.741 (0.75), 1.754 (1.29), 1.768 (1.40), 1.780 (1.32), 1.814 (0.72), 2.149 (6.09), 2.185 (4.76), 2.204 (7.72), 2.222 (3.48), 2.248 (0.41 ), 3.547 (0.97), 3.584 (1.69), 3.698 (2.95), 7.234 (1.33), 7.253 (2.63), 7.272 (1.78), 7.349 (1.71 ), 7.368 (2.91 ), 7.386 (1 .57), 7.431 (4.72), 7.451 (3.97), 7.485 (5.40), 7.501 (8.92), 7.508 (6.68), 7.520 (16.00), 7.532 (7.56), 7.699 (0.61 ), 7.834 (2.92), 7.839 (2.51 ), 7.856 (4.20), 7.861 (3.76), 7.930 (7.74), 7.952 (4.78), 8.832 (1.79), 8.847 (3.30), 8.860 (1.53).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1 1.95 (br. s, 1 H), 8.85 (t, 2H), 7.94 (d, 1 H), 7.85 (dd, 1 H), 7.72 (br. s, 1 H), 7.56-7.42 (m, 7H), 7.37 (t, 1 H), 7.25 (t, 1 H), 3.78-3.50 (m, 3H), 2.24-2.09 (m, 5H), 1.85-1.71 (m, 1 H), 1.70-1.57 (m, 1 H), 1.54-1.27 (m, 2H).
Beispiel 240
(+/-)-6-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino
hexansäure (Racemat)

Zu einer Lösung aus (+/-)-ie/f-Butyl-6-{[(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-5- (2-chlorphenyl)-5-methylhexanoat (866 mg,„1.36 mmol", nicht reinheitskorrigiert, erste Mischfraktion aus Beispiel 305A) in Dichlormethan (10 ml) wurde TFA (1.0 ml, 14 mmol) gegeben, und das Gemisch wurde zwei Tage bei RT stehen gelassen. Anschließend wurde das Gemisch eingeengt und mehrmals mit Dichlormethan versetzt und wieder eingeengt. Der Rückstand wurde in Aceto- nitril gelöst und mittels präparativer HPLC (Methode 16) gereinigt. Es wurden drei Fraktionen eingeengt. Nach Trocknen im Vakuum wurden aus der ersten Fraktion 189 mg (100% Reinheit) einer ersten Charge des de-chlorierten Produktes ((+/-)-6-{[(6-Brom-3-methyl-2-phenylchinolin-4- yl)carbonyl]amino}-5-methyl-5-phenylhexansäure, siehe Beispiel 243) erhalten. Aus der dritten Fraktion wurden 282 mg (96% Reinheit) einer ersten Charge der Titelverbindung erhalten. Die mittlere Fraktion wurde erneut mittels präparativer HPLC (Methode 26) gereinigt. Nach Einengen der betreffenden Zielfraktionen und Trocknen im Vakuum wurden 61 mg (100% Reinheit) einer zweiten Charge des de-chlorierten Produktes ((+/-)-6-{[(6-Brom-3-methyl-2-phenylchinolin-4- yl)carbonyl]amino}-5-methyl-5-phenylhexansäure, siehe Beispiel 243) erhalten. Außerdem wurden 67 mg (100% Reinheit, siehe Analytik) einer zweiten Charge der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.12 min; MS (ESIpos): m/z = 579/581 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 0.008 (2.57), 1.047 (0.92), 1.066 (1.29), 1.079 (1.36), 1.098 (1.09), 1.1 10 (0.75), 1.315 (1.08), 1.335 (1.35), 1.347 (1.37), 1.366 (1.1 1 ), 1.391 (1.30), 1.529 (16.00), 1.600 (1.04), 1.61 1 (1.1 1 ), 1.633 (1.91 ), 1.644 (1.75), 1.666 (1.15), 1.676 (0.94), 2.085 (10.08), 2.138 (5.60), 2.156 (9.00), 2.175 (4.42), 2.282 (0.46), 2.327 (0.47), 2.366 (0.42), 2.397 (0.92), 2.418 (1.50), 2.450 (0.85), 3.666 (1.69), 3.678 (1.84), 3.698 (2.02), 3.712 (1.97), 3.852 (1.49), 4.292 (1.78), 4.309 (1.97), 4.326 (1.79), 4.343 (1.64), 5.754 (8.27), 7.242 (1.04), 7.260 (2.63), 7.279 (2.41 ), 7.292 (2.38), 7.310 (3.20), 7.327 (1.59), 7.413 (3.43), 7.432 (6.46),
7.451 (3.16), 7.474 (0.83), 7.486 (2.00), 7.500 (6.53), 7.507 (5.67), 7.518 (14.80), 7.524 (13.14), 7.680 (0.60), 7.829 (2.76), 7.833 (2.56), 7.851 (4.25), 7.856 (4.03), 7.925 (7.72), 7.947 (4.86), 8.590 (1.85), 8.605 (3.26), 8.619 (1.86).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 12.01 (br. s, 1 H), 8.62 (t, 1 H), 7.93 (d, 1 H), 7.84 (dd, 1 H), 7.67 (br. s, 1 H), 7.56-7.39 (m, 7H), 7.35-7.21 (m, 2H), 4.31 (dd, 1 H), 3.69 (dd, 1 H), 2.47- 2.35 (m, 1 H), 2.24-2.02 (m, 5H), 1.64 (td, 1 H), 1.53 (s, 3H), 1.43-1.25 (m, 1 H), 1.15-0.99 (m, 1 H).
Trennung der Enantiomere:
Die Titelverbindung (247 mg) wurde in Methanol (15 ml) gelöst und mittels praparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 241 und 242) [Säule: Daicel Chiral- cel IC, 5 m, 250 mm x 20 mm; Fluss: 80 ml/min; Injektion: 0.30 ml; Eluent: 25% Ethanol / 75% Kohlendioxid; Laufzeit 19 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt, und der jeweilige Rückstand wurde im Vakuum getrocknet.
Beispiel 241
(-)-6-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-5-(2-chlorphenyl)-5-methyl- hexansäure (Enantiomer 1 )
Bei der in Beispiel 240 beschriebenen Enantiomerentrennung wurden 66 mg (95% Reinheit, ee- Wert >99%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -27.7°, 589 nm, c = 0.35 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.13 min; MS (ESIpos): m/z = 579/581 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.048 (0.93), 1.065 (1.23), 1.077 (1.29), 1.095 (1.07), 1.233 (0.55), 1.314 (1.06), 1.332 (1.34), 1.345 (1.33), 1.363 (1.09), 1.390 (1.60), 1.527 (16.00), 1.601 (1.01 ), 1.610 (1.08), 1.633 (1.83), 1.642 (1.71 ), 1.665 (1.04), 2.133 (4.41 ), 2.151 (5.76), 2.278 (0.54), 2.391 (1.00), 2.414 (1.60), 2.422 (1.59), 2.447 (0.93), 3.667 (1.35), 3.679 (1.46), 3.700 (1.57), 3.712 (1.44), 4.289 (1.68), 4.306 (1.85), 4.322 (1.69), 4.340 (1.55), 7.241 (1.11 ), 7.259 (2.75), 7.278 (2.56), 7.290 (2.48), 7.309 (3.37), 7.326 (1.71 ), 7.343 (0.60), 7.41 1 (3.64), 7.431 (6.51 ), 7.451 (3.18), 7.472 (0.93), 7.498 (6.58), 7.505 (5.67), 7.517 (14.29), 7.523 (13.42), 7.672 (0.63), 7.826 (2.64), 7.848 (4.09), 7.923 (6.70), 7.945 (4.28), 8.596 (1.73), 8.610 (3.12), 8.624 (1.78).
Beispiel 242
(+)-6-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-5-(2-chlorphenyl)-5-methyl- hexan säure (Enantiomer 2)
Bei der in Beispiel 240 beschriebenen Enantiomerentrennung wurden 60 mg (100% Reinheit, ee-Wert >99%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = +25.7°, 589 nm, c = 0.35 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.12 min; MS (ESIpos): m/z = 579/581 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (2.05), 0.008 (1.72), 1.077 (1.24), 1.1 13 (0.92), 1.232 (0.71 ), 1.332 (1.25), 1.527 (16.00), 1.61 1 (1.06), 1.633 (1.68), 1.664 (0.94), 2.149 (4.91 ), 2.277 (0.62), 2.327 (0.64), 2.387 (1.03), 2.414 (1.57), 2.443 (0.98), 2.670 (0.47), 3.540 (1.21 ), 3.668 (1.34), 3.680 (1.37), 3.701 (1.51 ), 4.288 (1.61 ), 4.305 (1.81 ), 4.322 (1.67), 4.338 (1.52), 7.241 (1.12), 7.259 (2.88), 7.278 (2.66), 7.291 (2.48), 7.308 (3.45), 7.326 (1.62), 7.41 1 (3.37), 7.431 (6.32), 7.451 (3.15), 7.472 (1.00), 7.484 (2.24), 7.498 (6.98), 7.505 (5.98), 7.516 (14.63), 7.523 (13.31 ), 7.667 (0.64), 7.825 (3.06), 7.830 (2.78), 7.848 (4.56), 7.853 (4.27), 7.923 (8.71 ), 7.945 (5.48), 8.600 (1.77), 8.615 (2.99), 8.630 (1.66).
Beispiel 243
(+/-)-6-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-5-methyl-5-phenylhexansäure (Racemat)
Wie in Beispiel 240 beschrieben, wurden 189 mg (100% Reinheit, siehe Analytik) einer ersten Charge der Titelverbindung und 61 mg (100% Reinheit) einer zweiten Charge der Titelverbindung erhalten.
LC-MS (Methode 1 ): Rt = 2.07 min; MS (ESIpos): m/z = 545/547 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.150 (0.16), 0.007 (1.24), 0.146 (0.16), 1.141 (0.62), 1.160 (0.85), 1.173 (0.93), 1.192 (0.80), 1.204 (0.51 ), 1.334 (0.82), 1.353 (1.06), 1.366 (1.17), 1.391 (16.00), 1.529 (0.36), 1.586 (0.62), 1.597 (0.70), 1.619 (1.29), 1.629 (1.23), 1.651 (0.85), 1.661 (0.68), 1.774 (0.75), 1.784 (0.82), 1.806 (1.18), 1.816 (1.12), 1.838 (0.58), 2.085 (1.70), 2.1 18 (4.12), 2.136 (6.68), 2.155 (3.41 ), 2.331 (0.27), 2.346 (0.24), 2.365 (0.43), 2.669 (0.18), 2.709 (0.18), 3.169 (2.40), 3.538 (1.47), 3.551 (1.61 ), 3.571 (1.97), 3.584 (1.97), 3.655 (1.18), 3.833 (1.50), 3.850 (1.59), 3.866 (1.33), 3.883 (1.21 ), 3.964 (0.23), 4.527 (0.14), 5.753 (0.28), 7.196 (0.88), 7.213 (2.20), 7.231 (1.49), 7.325 (2.43), 7.344 (4.84), 7.363 (2.95), 7.412 (6.54),
7.431 (4.28), 7.473 (0.54), 7.485 (1.44), 7.500 (4.46), 7.507 (3.68), 7.518 (8.57), 7.528 (8.40), 7.570 (0.34), 7.588 (0.28), 7.667 (0.57), 7.827 (1.92), 7.833 (1.76), 7.850 (2.94), 7.855 (2.81 ), 7.928 (5.42), 7.950 (3.54), 8.598 (1.29), 8.613 (2.28), 8.628 (1.26).
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1 1.97 (br. s, 1 H), 8.61 (t, 1 H), 7.94 (d, 1 H), 7.84 (dd, 1 H), 7.67 (br. s, 1 H), 7.59-7.46 (m, 5H), 7.45-7.39 (m, 2H), 7.34 (t, 2H), 7.22 (t, 1 H), 3.86 (dd, 1 H), 3.56 (dd, 1 H), 2.25-1.98 (m, 5H), 1.81 (td, 1 H), 1.62 (td, 1 H), 1.45-1.29 (m, 1 H), 1.39 (s, 3H), 1.25-1.09 (m, 1 H).
Trennung der Enantiomere:
Die Titelverbindung (160 mg) wurde in Methanol (20 ml) gelöst und mittels praparativer SFC an chiraler Phase in die Enantiomere getrennt (siehe Beispiele 244 und 245) [Säule: Daicel Chiral- pak AD, 250 mm x 20 mm; Fluss: 80 ml/min; Injektion: 0.50 ml; Eluent: 12% Ethanol / 88% Kohlendioxid; Laufzeit 22 min, isokratisch]. Die vereinigten Zielfraktionen wurden eingeengt, und der jeweilige Rückstand wurde im Vakuum getrocknet.
Beispiel 244
(-)-6-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-5-methyl-5-phenylhexansäure (Enantiomer 1)
Bei der in Beispiel 243 beschriebenen Enantiomerentrennung wurden 56 mg (100% Reinheit, ee-Wert 99%) der Titelverbindung als früher eluierendes Enantiomer erhalten.
[a]D 20 = -18.7°, 589 nm, c = 0.40 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.07 min; MS (ESIpos): m/z = 545/547 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: -0.008 (1.29), 0.008 (1.06), 1.030 (0.82), 1.045 (0.85), 1.140 (0.68), 1.160 (0.92), 1.172 (1.00), 1.191 (0.83), 1.333 (0.83), 1.352 (1.07), 1.390 (16.00), 1.585 (0.67), 1.597 (0.76), 1.618 (1.35), 1.629 (1.28), 1.650 (0.90), 1.661 (0.71 ), 1.773 (0.78), 1.782 (0.87), 1.805 (1.23), 1.814 (1.18), 1.837 (0.61 ), 1.846 (0.52), 2.072 (1.18), 2.1 15 (4.26), 2.133 (6.73), 2.152 (3.32), 2.730 (1.67), 2.889 (1.91 ), 3.538 (1.13), 3.551 (1.24), 3.572 (1.45), 3.585 (1.34), 3.833 (1.37), 3.850 (1.52), 3.866 (1.24), 3.883 (1.12), 7.196 (1.02), 7.213 (2.47), 7.231 (1.65), 7.324 (2.71 ), 7.344 (5.30), 7.363 (3.21 ), 7.412 (6.82), 7.431 (4.23), 7.472 (0.60), 7.484 (1.69), 7.499 (5.05), 7.506 (4.13), 7.517 (9.35), 7.527 (9.32), 7.545 (1.74), 7.669 (0.66), 7.826 (2.19), 7.831 (1.97), 7.848 (3.28), 7.853 (3.08), 7.927 (6.46), 7.949 (4.33), 8.602 (1.35), 8.617 (2.32), 8.632 (1.27).
Beispiel 245
(+)-6-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-5-methyl-5-phenylhexansäure (Enantiomer 2)
Bei der in Beispiel 243 beschriebenen Enantiomerentrennung wurden 35 mg (100% Reinheit, ee-Wert 94%) der Titelverbindung als später eluierendes Enantiomer erhalten.
[a]D 20 = +18.0°, 589 nm, c = 0.35 g/100 ml, Methanol
LC-MS (Methode 1 ): Rt = 2.07 min; MS (ESIpos): m/z = 545/547 [M+H]+
Ή-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.141 (0.68), 1.161 (0.94), 1.173 (1.04), 1.192 (0.85), 1.334 (0.85), 1.353 (1.12), 1.391 (16.00), 1.587 (0.64), 1.597 (0.75), 1.619 (1.37), 1.629 (1.29), 1.651 (0.91 ), 1.662 (0.72), 1.784 (0.88), 1.806 (1.25), 1.816 (1.20), 1.839 (0.64), 2.1 18 (4.31 ), 2.137 (6.84), 2.155 (3.37), 2.731 (0.94), 2.890 (1.03), 3.537 (1.19), 3.551 (1.22), 3.571 (1.40), 3.585 (1.31 ), 3.833 (1.37), 3.850 (1.49), 3.866 (1.25), 3.883 (1.15), 7.196 (1.01 ), 7.214 (2.46), 7.232 (1.68), 7.325 (2.70), 7.344 (5.36), 7.363 (3.26), 7.412 (6.97), 7.432 (4.22), 7.473 (0.59), 7.485 (1.59), 7.500 (4.92), 7.506 (4.04), 7.518 (9.53), 7.528 (9.42), 7.667 (0.68), 7.827 (2.12), 7.831 (1.97), 7.849 (3.18), 7.854 (3.09), 7.927 (6.17), 7.949 (4.02), 8.598 (1.37), 8.613 (2.42), 8.628 (1.31 ), 1 1.939 (1.52).
Beispiel 246
1 -[2-{[(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-1 -(2-methoxyphenyl)ethyl]- pyrrolidin-3-carbonsäure
Zu einem Gemisch aus 6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure (75 mg, 219 μηηοΙ, darstellbar gemäß WO 2016 146602 A1 , S. 51 , Beispiel 3A) in DMF (2 ml) wurden HATU (125 mg, 329 μηιοΙ) und DIPEA (1 10 μΙ, 660 μηιοΙ) gegeben und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde 1-[2-Amino-1-(2-methoxyphenyl)ethyl]pyrrolidin-3-carbonsäure (102 mg, 85% Reinheit, 329 μηιοΙ, CAS-RN 886363-84-4, kommerziell verfügbar), gelöst in DMF (1 ml) hinzugegeben, und das Gemisch wurde 3 h bei 60 °C gerührt. Anschließend wurde das Gemisch mit Ethylacetat und Wasser (jeweils 20 ml) versetzt und geschüttelt. Nach Phasentren- nung wurde die wässrige Phase zweimal mit Ethylacetat (20 ml) extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Nat-
riumsulfat getrocknet, filtriert und eingeengt. Beim Einengen fiel ein Feststoff aus, welcher abfiltriert wurde. Das Filtrat wurde eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen und mittels Flash-Säulenchromatographie (50 g Kieselgel Biotage Snap-Cartridge KP- Sil, Cyclohexan / Ethylacetat 85:15, Isolera One) vorgereinigt. Die vereinigten Zielfraktionen wurden eingeengt, und der Rückstand wurde mittels praparativer HPLC (Methode 16) nachgereinigt. Anschließend wurde erneut mittels praparativer HPLC (Methode 22) nachgereinigt. Anschließend wurde erneut mittels praparativer HPLC (Methode 25) nachgereinigt. Es wurden 4 mg (90% Reinheit, 3% d. Th.) der Titelverbindung erhalten.
LC-MS (Methode 2): Rt = 0.80 & 0.82 min; MS (ESIpos): m/z = 588/590 [M+H]+
1H-NMR (400 MHz, DMSO-d6) δ [ppm]: 1.236 (0.85), 1.886 (1.62), 1.899 (1.67), 2.167 (12.14), 2.183 (2.43), 2.256 (0.64), 2.328 (0.71 ), 2.366 (0.76), 2.403 (0.48), 2.421 (0.53), 2.451 (0.74), 2.569 (2.03), 2.589 (1.30), 2.637 (0.52), 2.670 (0.72), 2.710 (0.41 ), 2.765 (2.17), 2.774 (4.88), 2.834 (1.19), 2.872 (0.89), 2.905 (1.19), 2.939 (2.25), 2.958 (5.33), 3.724 (0.92), 3.783 (16.00), 3.910 (0.82), 4.181 (1.03), 4.195 (1.76), 4.208 (0.91 ), 4.256 (0.68), 6.928 (1.16), 6.947 (2.36), 6.965 (1.35), 7.003 (2.1 1 ), 7.023 (2.44), 7.232 (1.32), 7.250 (2.08), 7.270 (0.95), 7.337 (0.85), 7.354 (1.91 ), 7.370 (1.78), 7.385 (0.70), 7.474 (0.63), 7.487 (1.71 ), 7.501 (4.65), 7.506 (4.14), 7.520 (8.73), 7.531 (9.03), 7.795 (1.46), 7.836 (2.96), 7.860 (3.31 ), 7.926 (4.22), 7.948 (2.61 ), 8.584 (1.09), 8.599 (1.48), 8.614 (0.99).
B. Bewertung der pharmakologischen Wirksamkeit Die pharmakologische Aktivität der erfindungsgemäßen Verbindungen kann durch in vitro- und in v/Vo-Untersuchungen, wie sie dem Fachmann bekannt sind, nachgewiesen werden. Die nachfolgenden Anwendungsbeispiele beschreiben die biologische Wirkung der erfindungsgemäßen Verbindungen, ohne die Erfindung auf diese Beispiele zu beschränken.
Abkürzungen und Akronyme:
CRTH2 Chemoattractant Receptor-homologous molecule expressed on T-Helper type 2 cells
DMEM Dulbecco's modified Eagle's medium
DMSO Dimethylsulfoxid
DP PGD2-Rezeptor
EC50 halbmaximale effektive Konzentration
Em Emission
EP PGE2-Rezeptor
Ex Exzitation
Fa. Firma (Bezugsquelle)
FCS fötales Kälberserum
FP PGF2a-Rezeptor
HEPES 2-[4-(2-Hydroxyethyl)piperazin-1-yl]ethansulfonsäure
I C50 halbmaximale inhibitorische Konzentration
IP PGI2-rezeptor
Lit. Literatur(stelle)
MES 2-(/V-Morpholino)ethansulfonsäure
Pen/Strep Penicillin/Streptomycin
PGD2 Prostaglandin D2
PGE2 Prostaglandin E2
PGF2a Prostaglandin F2a
PGI2 Prostaglandin 12
TC tissue culture
TP Thromboxan A2-Rezeptor
Tris Tris(hydroxymethyl)aminomethan
v/v Volumen zu Volumen-Verhältnis (einer Lösung)
w/w Gewicht zu Gewicht-Verhältnis (einer Lösung)
B-1. In vitro-Jest auf Hemmung der humanen FP-Rezeptor-Aktivität
Für die Charakterisierung von Testsubstanzen auf FP-Antagonismus wurde der PGF2a- induzierte Calcium-Flux in FP-exprimierenden CHEM1 -Zellen (Millipore, HTS093C) verwendet. 3000 Zellen in 30 μΙ Vollmedium [DMEM F12, 10% FCS, 1.35 mM Natriumpyruvat, 20 mM HEPES, 4 mM GlutaMAX™, 2% Natriumbicarbonat, 1 % Pen/Strep, 1 % 100x nicht-essentielle Aminosäuren] werden je Vertiefung einer 384-Multititerplatte (Fa. Greiner, TC-Platte, schwarz mit klarem Boden) ausgesät und bei 33°C, 5% CO2 für 24 Stunden inkubiert. Vor der Messung wird das Medium durch 30 μΙ Fluo-8 AM-Beladungspuffer [Calcium-freie Tyrode (130 mM NaCI, 5 mM KCl, 20 mM HEPES, 1 mM MgCI2, 4.8 mM NaHCOs, pH 7.4), 2 mM CaCI2, 6.3 mM Probenecid, 5 μΜ Fluo-8 AM, 0.01 12% Pluronic®] ersetzt und bei 37°C, 5% C02 für 30 Minuten inkubiert. Die Testsubstanz wird in DMSO in verschiedenen Konzentrationen als Dosiswirkungskurve vorbereitet (Startkonzentration 10 mM, Verdünnungsfaktor 3.16) und 1 :50 mit Calcium-freier Tyrode, 2 mM CaCb, 0.002% SmartBlock (Fa. CANDOR Bioscience GmbH) vorverdünnt. 10 μΙ der vor- verdünnten Substanzlösung werden zu den Fluo-8-beladenen Zellen gegeben und bei 37°C, 5% CO2 für 10 Minuten inkubiert. Der FP-Rezeptor wird durch Zugabe von 40 μΙ 2 nM (finale Konzentration) PGF2a in Calcium-freier Tyrode, 2 mM CaCb, 0.002% SmartBlock aktiviert, und der Calcium-Flux wird durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden in einem Fluoreszenzmessgerät (FLIPR Tetra®, Molecular Devices) bestimmt.
ln der folgenden Tabelle 1 sind für individuelle Ausführungsbeispiele der Erfindung die IC50- Werte aus diesem Assay aufgeführt (zum Teil als Mittelwerte aus mehreren unabhängigen Einzelbestimmungen):
Tabelle 1
FP-Rezeptor- FP-Rezeptor- FP-Rezeptor-
Bsp. Bsp. Bsp.
aktivität aktivität aktivität Nr. Nr. Nr.
IC50 [Mmol/L] IC50 [Mmol/L] IC50 [Mmol/L]
1 0, 13 30 0,34 59 0,39
2 0, 16 31 0,068 60 0,12
3 0,27 32 0,15 61 0,23
4 0,031 33 0,19 62 0,043
5 0,037 34 0,13 63 0,34
6 0,032 35 0,19 64 0,34
7 0,024 36 0,81 65 0,22
8 0, 13 37 0,16 66 0,50
9 0,0072 38 0,047 67 0,37
10 0,32 39 0,14 68 >1 ,0
1 1 0,47 40 0,034 69 0,048
12 0, 18 41 0,13 70 0,032
13 0,094 42 0,12 71 0,036
14 0,053 43 0,14 72 0,045
15 0, 19 44 0,19 73 0,086
16 0, 17 45 0,60 74 0,079
17 0,26 46 0,23 75 0,069
18 0, 12 47 0,043 76 0,023
19 0, 15 48 0,033 77 0,085
20 0,097 49 0,069 78 0,10
21 0, 17 50 0,079 79 0,14
22 0,076 51 0,042 80 0,051
23 0,53 52 0,071 81 0,19
24 0,58 53 0,18 82 0,22
25 >1 ,0 54 0,50 83 0,28
26 0,044 55 0,078 84 0,016
27 0,053 56 0,021 85 0,016
28 0, 10 57 0,21 86 0,24
29 0, 14 58 0,0097 87 0,023
FP-Rezeptor- FP-Rezeptor- FP-Rezeptor-
Bsp. Bsp. Bsp.
aktivität aktivität aktivität Nr. Nr. Nr.
ICso [Mmol/L] ICso [Mmol/L] ICso [Mmol/L]
88 0,019 121 0,010 154 0,015
89 0,027 122 0,46 155 0,060
90 0,47 123 0,01 1 156 0,0080
91 0,46 124 0,0053 157 0,0037
92 0,32 125 0,097 158 0,0035
93 0,080 126 0,039 159 0,024
94 0,096 127 0,025 160 0,013
95 0,084 128 0,1 1 161 0,0082
96 0,023 129 0,13 162 0,033
97 0, 12 130 0,32 163 0,0087
98 0,0090 131 0,099 164 0,015
99 0,028 132 0,032 165 0,0066
100 0,020 133 0,047 166 0,0061
101 0,032 134 0,80 167 0,026
102 0,46 135 0,013 168 0,0036
103 0,29 136 0,013 169 0,026
104 >1 ,0 137 0,44 170 0,1 1
105 0,092 138 0,042 171 0,01 1
106 0,28 139 0,028 172 0,0032
107 0,051 140 >1 ,0 173 0,001 1
108 0,078 141 0,024 174 0,019
109 0,075 142 0,015 175 0,015
110 0,075 143 0,0068 176 0,012
11 1 0,089 144 0,13 177 0,019
112 0,092 145 0,049 178 0,0065
113 0,062 146 >1 ,0 179 0,0041
114 0,018 147 >1 ,0 180 0,0082
115 0,031 148 0,042 181 0,0092
116 0,074 149 >1 ,0 182 0,0072
117 0,24 150 0,96 183 0,023
118 0,28 151 0,0078 184 0,0077
119 0, 15 152 0,0079 185 0,0039
120 0,030 153 0,0090 186 0,017
FP-Rezeptor- FP-Rezeptor- FP-Rezeptor-
Bsp. Bsp. Bsp.
aktivität aktivität aktivität Nr. Nr. Nr.
ICso [Mmol/L] ICso [Mmol/L] ICso [Mmol/L]
187 0,036 209 0,0018 231 0,022
188 0,054 210 0,0061 232 0,0088
189 0,062 21 1 0,0017 233 0,025
190 0,067 212 0,0066 234 0,0072
191 0, 10 213 0,0028 235 0,032
192 0,042 214 0,0044 236 0,0020
193 0, 15 215 0,0054 237 0,0052
194 0,0079 216 0,0050 238 0,0043
195 0,0080 217 0,019 239 0,010
196 0,012 218 0,013 240 0,024
197 0,021 219 0,010 241 0,068
198 0,40 220 0,0080 242 0,016
199 0,025 221 0,0024 243 0,031
200 0,0036 222 0,00096 244 0,020
201 0,0012 223 0,012 245 0,063
202 0,0062 224 0,10 246 0,094
203 0,0076 225 0,0047 247 0,0072
204 0,0037 226 0,10 248 0,013
205 0,022 227 0,0016 249 0,0028
206 0,013 228 0,0042 250 0,017
207 0,010 229 0,0027 251 0,015
208 0,01 1 230 0,049 252 0,014
B-2. In vitro FP-Rezeptorbindunq-lnhibitionstest
Für den FP-Rezeptorbindungstest werden humane rekombinante Prostanoid-FP-Rezeptoren, exprimiert in HEK293-Zellen, in modifiziertem MES-Puffer, pH 6.0, verwendet. Die Durchführung dieses Tests wird kommerziell angeboten (Fa. Eurofins Panlabs, Katalog #268510). 80 μg Membran werden mit 1 nM [3H]-PGF2a für 60 Minuten bei 25°C inkubiert. Die Menge an Membranprotein kann von Lot zu Lot variieren und wird bei Bedarf angepasst. Unspezifische Bindung wird in Anwesenheit von 1 μΜ Cloprostenol bestimmt. Die Membranen werden gefiltert, gewaschen und dann vermessen, um die spezifische Bindung von [3H]-PGF2a zu bestimmen. Sub- stanzen werden auf inhibitorische Aktivität bei einer Konzentration von 10 μΜ oder in Form einer Dosiswirkungskurve getestet [Lit: Abramovitz et al., J. Biol. Chem. 1994, 269 (4): 2632].
B-3. In vitro CRTH2-Rezeptorbindung-lnhibitionstest
Für diesen Test werden humane rekombinante Prostanoid-CRTH2-Rezeptoren, exprimiert in CHO-K1 -Zellen, in modifiziertem Tris-HCI-Puffer, pH 7.4, verwendet. Die Durchführung dieses Tests wird kommerziell angeboten (Fa. Eurofins Panlabs, Katalog #268030). 4 μg Membran wer- den mit 1 nM [3H]-PGD2 für 120 Minuten bei 25°C inkubiert. Die Menge an Membranprotein kann von Lot zu Lot variieren und wird bei Bedarf angepasst. Unspezifische Bindung wird in Anwesenheit von 1 μΜ PGD2 bestimmt. Die Membranen werden gefiltert, gewaschen und dann vermessen, um die spezifische Bindung von [3H]-PGD2 zu bestimmen. Substanzen werden auf inhibitorische Aktivität bei einer Konzentration von 10 μΜ oder in Form einer Dosiswirkungskurve getestet [Lit.: Sugimoto et al., J. Pharmacol. Exp. Ther. 2003, 305 (1 ): 347].
B-4. In vitro DP-Rezeptorbindunq-lnhibitionstest
Für diesen Test werden humane rekombinante Prostanoid-DP-Rezeptoren, exprimiert in Chem-1- Zellen, in modifiziertem HEPES-Puffer, pH 7.4, verwendet. Die Durchführung dieses Tests wird kommerziell angeboten (Fa. Eurofins Panlabs, Katalog #268060). 10 μg Membran werden mit 2 nM [3H]-PGD2 für 120 Minuten bei 25°C inkubiert. Die Menge an Membranprotein kann von Lot zu Lot variieren und wird bei Bedarf angepasst. Unspezifische Bindung wird in Anwesenheit von 1 μΜ PGD2 bestimmt. Die Membranen werden gefiltert, gewaschen und dann vermessen, um die spezifische Bindung von [3H]-PGD2 zu bestimmen. Substanzen werden auf inhibitorische Aktivität bei einer Konzentration von 10 μΜ oder in Form einer Dosiswirkungskurve getestet [Lit.: Wright et al., Br. J. Pharmacol. 1998, 123 (7): 1317; Sharif et al., Br. J. Pharmacol. 2000, 131 (6): 1025].
B-5. In vitro EP1 -Rezeptorbindung-Inhibitionstest
Für diesen Test werden humane rekombinante Prostanoid-EP 1 -Rezeptoren, exprimiert in HEK293-Zellen, in modifiziertem MES-Puffer, pH 6.0, verwendet. Die Durchführung dieses Tests wird kommerziell angeboten (Fa. Eurofins Panlabs, Katalog #2681 10). 14 μg Membran werden mit 1 nM [3H]-PGE2 für 60 Minuten bei 25°C inkubiert. Die Menge an Membranprotein kann von Lot zu Lot variieren und wird bei Bedarf angepasst. Unspezifische Bindung wird in Anwesenheit von 10 μΜ PGE2 bestimmt. Die Membranen werden gefiltert, gewaschen und dann vermessen, um die spezifische Bindung von [3H]-PGE2 zu bestimmen. Substanzen werden auf inhibitorische Aktivität bei einer Konzentration von 10 μΜ oder in Form einer Dosiswirkungskurve getestet [Lit.: Abra- movitz et al., Biochim. Biophys. Acta 2000, 1483 (2): 285; Funk et al., J. Biol. Chem. 1993, 268 (35): 26767].
B-6. In vitro EP2-Rezeptorbindung-lnhibitionstest
Für diesen Test werden humane rekombinante Prost.anoid-EP2-Rezept.oren, exprimiert in HEK293-Zellen, in modifiziertem MES/KOH-Puffer, pH 6.0, verwendet. Die Durchführung dieses
Tests wird kommerziell angeboten (Fa. Eurofins Panlabs, Katalog #268200). 25 mg/ml Membran werden mit 4 nM [3H]-PGE2 für 120 Minuten bei 25°C inkubiert. Die Menge an Membranprotein kann von Lot zu Lot variieren und wird bei Bedarf angepasst. Unspezifische Bindung wird in Anwesenheit von 10 μΜ PGE2 bestimmt. Die Membranen werden gefiltert, gewaschen und dann vermessen, um die spezifische Bindung von [3H]-PGE2 zu bestimmen. Substanzen werden auf inhibitorische Aktivität bei einer Konzentration von 10 μΜ oder in Form einer Dosiswirkungskurve getestet [Lit. : Bastien et al., J. Biol. Chem. 1994, 269 (16): 1 1873; Boie et al., Eur. J. Pharmacol. 1997, 340 (2-3): 227].
B-7. In vitro EP3-Rezeptorbindunq-lnhibitionstest
Für diesen Test werden humane rekombinante Prost.anoid-EP3-Rezept.oren, exprimiert in HEK293-Zellen, in modifiziertem MES-Puffer, pH 6.0, verwendet. Die Durchführung dieses Tests wird kommerziell angeboten (Fa. Eurofins Panlabs, Katalog #268310). 3 μg Membran werden mit 0.5 nM [3H]-PGE2 für 120 Minuten bei 25°C inkubiert. Die Menge an Membranprotein kann von Lot zu Lot variieren und wird bei Bedarf angepasst. Unspezifische Bindung wird in Anwesen- heit von 10 μΜ PGE2 bestimmt. Die Membranen werden gefiltert, gewaschen und dann vermessen, um die spezifische Bindung von [3H]-PGE2 zu bestimmen. Substanzen werden auf inhibitorische Aktivität bei einer Konzentration von 10 μΜ oder in Form einer Dosiswirkungskurve getestet [Lit.: Schmidt et al., Eur. J. Biochem. 1995, 228 (1 ): 23].
B-8. In vitro EP4-Rezeptorbindung-lnhibitionstest
Für diesen Test werden humane rekombinante Prost.anoid-EP4-Rezept.oren, exprimiert in Chem- 1 -Zellen, in modifiziertem MES-Puffer, pH 6.0, verwendet. Die Durchführung dieses Tests wird kommerziell angeboten (Fa. Eurofins Panlabs, Katalog #268420). 3 μg Membran werden mit 1 nM [3H]-PGE2 für 120 Minuten bei 25°C inkubiert. Die Menge an Membranprotein kann von Lot zu Lot variieren und wird bei Bedarf angepasst. Unspezifische Bindung wird in Anwesenheit von 10 μΜ PGE2 bestimmt. Die Membranen werden gefiltert, gewaschen und dann vermessen, um die spezifische Bindung von [3H]-PGE2 zu bestimmen. Substanzen werden auf inhibitorische Aktivität bei einer Konzentration von 10 μΜ oder in Form einer Dosiswirkungskurve getestet [Lit.: Davis et al., Br. J. Pharmacol. 2000, 130 (8): 1919].
B-9. In vitro IP-Rezeptorbindunq-lnhibitionstest
Für diesen Test werden humane rekombinante Prostanoid-IP-Rezeptoren, exprimiert in HEK293-Zellen, in modifiziertem HEPES-Puffer, pH 6.0, verwendet. Die Durchführung dieses Tests wird kommerziell angeboten (Fa. Eurofins Panlabs, Katalog #268600). 15 μg Membran werden mit 5 nM [3H]-lloprost für 60 Minuten bei 25°C inkubiert. Die Menge an Membranprotein kann von Lot zu Lot variieren und wird bei Bedarf angepasst. Unspezifische Bindung wird in An- Wesenheit von 10 μΜ lloprost bestimmt. Die Membranen werden gefiltert, gewaschen und dann
vermessen, um die spezifische Bindung von [3H]-lloprost zu bestimmen. Substanzen werden auf inhibitorische Aktivität bei einer Konzentration von 10 μΜ oder in Form einer Dosiswirkungskurve getestet [Lit. : Armstrong et al., Br. J. Pharmacol. 1989, 97 (3): 657; Boie et al., J. Biol. Chem. 1994, 269 (16): 12173]. B-10. In vitro TP-Rezeptorbindung-lnhibitionstest
Für diesen Test werden humane rekombinante Prostanoid-TP-Rezeptoren, exprimiert in HEK- 293 EBNA-Zellen, in modifiziertem Tris/HCI-Puffer, pH 7.4, verwendet. Die Durchführung dieses Tests wird kommerziell angeboten (Fa. Eurofins Panlabs, Katalog #285510). 18.4 μg Membran werden mit 5 nM [3H]-SQ-29,548 für 30 Minuten bei 25°C inkubiert. Die Menge an Membran- protein kann von Lot zu Lot variieren und wird bei Bedarf angepasst. Unspezifische Bindung wird in Anwesenheit von 1 μΜ SQ-29,548 bestimmt. Die Membranen werden gefiltert, gewaschen und dann vermessen, um die spezifische Bindung von [3H]-SQ-29,548 zu bestimmen. Substanzen werden auf inhibitorische Aktivität bei einer Konzentration von 10 μΜ oder in Form einer Dosiswirkungskurve getestet [Lit.: Saussy Jr. et al., J. Biol. Chem. 1986, 261 : 3025; Hedberg et al., J. Pharmacol. Exp. Ther. 1988, 245: 786].
B-11. In vitro-Jest auf DP-Agonismus und -Antagonismus
Für die Charakterisierung von Testsubstanzen auf DP-Agonismus und -Antagonismus wurde der PGD2-induzierte Calcium-Flux in DP-exprimierenden CHEM1 -Zellen (Millipore, HTS091 C) verwendet: 3000 Zellen in 25 μΙ Vollmedium [DMEM, 4.5 g/l Glucose, 10% Hitze-inaktiviertes FCS, 1 % 100x nicht-essentielle Aminosäuren, 10 mM HEPES, 0.25 mg/ml Geneticin (G418), 100 U/ml Penicillin und Streptomycin] werden je Vertiefung einer 384-Multititerplatte (Fa. Greiner, TC-Platte, schwarz mit klarem Boden) ausgesät und bei 37°C / 5% CO2 für 24 Stunden inkubiert. Vor der Messung wird das Medium durch 30 μΙ Calcium-Farbstoff-Beladungspuffer (FLIPR Calcium Assay, Molecular Devices) ersetzt und bei 37°C / 5% CO2 für 60 Minuten inkubiert. Die Testsubstanz wird in DMSO in verschiedenen Konzentrationen als Dosiswirkungskurve vorbereitet (Startkonzentration 10 mM, Verdünnungsfaktor 3.16) und 1 :50 mit zum Beispiel Calcium-freier Tyrode (130 mM NaCI, 5 mM KCl, 20 mM HEPES, 1 mM MgCI2, 4.8 mM NaHCOs, pH 7.4) / 2 mM CaCI2 vorverdünnt. Für die DP-Agonismus-Messung werden in einem Fluoreszenzmessgerät (FLIPR Tetra®, Molecular Devices) 10 μΙ der vorverdünnten Substanzlösung zu den mit Calcium-Farbstoff beladenen Zellen gegeben und der Calcium-Flux durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt. Danach werden die Zellen bei 37°C / 5% CO2 für 10 Minuten inkubiert. Für die DP-Antagonismus-Messung wird der DP-Rezeptor im FLIPR Tetra® durch Zugabe von 20 μΙ -76 nM (2 x EC50, finale Konzentration) PGD2 in zum Beispiel Calcium- freier Tyrode / 2 mM CaC aktiviert und der Calcium-Flux durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt [Lit.: T. Matsuoka et al. (2000) Science 287: 2013-2017; S. Narumiya und G. A. Fitzgerald (2001 ) J. Clin. Invest. 108: 25-30].
B-12. In vitro-Jest auf EP1 -Agonismus und -Antagonismus
Für die Charakterisierung von Testsubstanzen auf EP1-Agonismus und -Antagonismus wurde der PGE2-induzierte Calcium-Flux in EP1-exprimierenden CHEM1-Zellen (Millipore, HTS099C) verwendet: 3000 Zellen in 25 μΙ Vollmedium [DMEM, 4.5 g/l Glucose, 10% Hitze-inaktiviertes FCS, 1 % 100x nicht-essentielle Aminosäuren, 10 mM HEPES, 0.25 mg/ml Geneticin (G418), 100 U/ml Penicillin und Streptomycin] werden je Vertiefung einer 384-Multititerplatte (Fa. Greiner, TC-Platte, schwarz mit klarem Boden) ausgesät und bei 37°C / 5% CO2 für 24 Stunden inkubiert. Vor der Messung wird das Medium durch 30 μΙ Calcium-Farbstoff-Beladungspuffer (FLIPR Calcium Assay, Molecular Devices) ersetzt und bei 37°C / 5% CO2 für 60 Minuten inkubiert. Die Testsubstanz wird in DMSO in verschiedenen Konzentrationen als Dosiswirkungskurve vorbereitet (Startkonzentration 10 mM, Verdünnungsfaktor 3.16) und 1 :50 mit zum Beispiel Calcium-freier Tyrode (130 mM NaCI, 5 mM KCl, 20 mM HEPES, 1 mM MgCI2, 4.8 mM NaHCOs, pH 7.4) / 2 mM CaCI2 vorverdünnt. Für die EP1-Agonismus-Messung werden in einem Fluoreszenzmessgerät (FLIPR Tetra®, Molecular Devices) 10 μΙ der vorverdünnten Substanzlösung zu den mit Calcium-Farbstoff beladenen Zellen gegeben und der Calcium-Flux durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt. Danach werden die Zellen bei 37°C / 5% CO2 für 10 Minuten inkubiert. Für die EP1 -Antagonismus-Messung wird der EP1-Rezeptor im FLIPR Tetra® durch Zugabe von 20 μΙ ~6 nM (2 x EC50, finale Konzentration) PGE2 in zum Beispiel Calcium- freier Tyrode / 2 mM CaC aktiviert und der Calcium-Flux durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt [Lit.: Y. Matsuoka et al. (2005) Proc. Natl. Acad. Sei. USA 102: 16066-16071 ; S. Narumiya und G. A. Fitzgerald (2001 ) J. Clin. Invest. 108: 25-30; K. Watanabe et al. (1999) Cancer Res. 59: 5093-5096].
B-13. In vitro-Jest auf EP2-Aqonismus und -Antagonismus
Für die Charakterisierung von Testsubstanzen auf EP2-Agonismus und -Antagonismus wurde der PGE2-induzierte Calcium-Flux in EP2-exprimierenden CHEM9-Zellen (Millipore, HTS185C) verwendet: 3000 Zellen in 25 μΙ Plattiermedium [DMEM, 4.5 g/l Glucose, 4 mM Glutamin, 10% Hitze-inaktiviertes FCS, 1 % 100x nicht-essentielle Aminosäuren, 10 mM HEPES, 100 U/ml Penicillin und Streptomycin] werden je Vertiefung einer 384-Multititerplatte (Fa. Greiner, TC-Platte, schwarz mit klarem Boden) ausgesät und bei 37°C / 5% CO2 für 24 Stunden inkubiert. Vor der Messung wird das Medium durch 30 μΙ Calcium-Farbstoff-Beladungspuffer (FLIPR Calcium Assay, Molecular Devices) ersetzt und bei 37°C / 5% CO2 für 60 Minuten inkubiert. Die Testsubstanz wird in DMSO in verschiedenen Konzentrationen als Dosiswirkungskurve vorbereitet (Startkonzentration 10 mM, Verdünnungsfaktor 3.16) und 1 :50 mit zum Beispiel Calcium-freier Tyrode (130 mM NaCI, 5 mM KCl, 20 mM HEPES, 1 mM MgCI2, 4.8 mM NaHCOs, pH 7.4) / 2 mM CaC vorverdünnt. Für die EP2-Agonismus-Messung werden in einem Fluoreszenzmessgerät (FLIPR Tetra®, Molecular Devices) 10 μΙ der vorverdünnten Substanzlösung zu den mit
Calcium-Farbstoff beladenen Zellen gegeben und der Calcium-Flux durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt. Danach werden die Zellen bei 37°C / 5% CO2 für 10 Minuten inkubiert. Für die EP2-Antagonismus-Messung wird der EP2- Rezeptor im FLIPR Tetra® durch Zugabe von 20 μΙ -22 nM (2 x EC50, finale Konzentration) PGE2 in zum Beispiel Calcium-freier Tyrode / 2 mM CaC aktiviert und der Calcium-Flux durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt [Lit. : C. R. Kennedy et al. (1999) Nat. Med. 5: 217-220; S. Narumiya und G. A. Fitzgerald (2001 ) J. Clin. In- vest. 108: 25-30; N. Yang et al. (2003) J. Clin. Invest. 1 1 1 : 727-735].
B-14. In vitro-Jest auf EP3-Aqonismus und -Antagonismus
Für die Charakterisierung von Testsubstanzen auf EP3-Agonismus und -Antagonismus wurde der PGE2-induzierte Calcium-Flux in EP3 (splice-Variante 6)-exprimierenden CHEM1-Zellen (Millipore, HTS092C) verwendet: 3000 Zellen in 25 μΙ Plattiermedium [DMEM, 4.5 g/l Glucose, 4 mM Glutamin, 10% Hitze-inaktiviertes FCS, 1 % 100x nicht-essentielle Aminosäuren, 10 mM HEPES, 100 U/ml Penicillin und Streptomycin] werden je Vertiefung einer 384-Multititerplatte (Fa. Greiner, TC-Platte, schwarz mit klarem Boden) ausgesät und bei 37°C / 5% CO2 für 24 Stunden inkubiert. Vor der Messung wird das Medium durch 30 μΙ Calcium-Farbstoff-Beladungspuffer (FLIPR Calcium Assay, Molecular Devices) ersetzt und bei 37°C / 5% CO2 für 60 Minuten inkubiert. Die Testsubstanz wird in DMSO in verschiedenen Konzentrationen als Dosiswirkungskurve vorbereitet (Startkonzentration 10 mM, Verdünnungsfaktor 3.16) und 1 :50 mit zum Beispiel Calcium-freier Ty- rode (130 mM NaCI, 5 mM KCl, 20 mM HEPES, 1 mM MgCI2, 4.8 mM NaHCOs, pH 7.4) / 2 mM CaC vorverdünnt. Für die EP3-Agonismus-Messung werden in einem Fluoreszenzmessgerät (FLIPR Tetra®, Molecular Devices) 10 μΙ der vorverdünnten Substanzlösung zu den mit Calcium- Farbstoff beladenen Zellen gegeben und der Calcium-Flux durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt. Danach werden die Zellen bei 37°C / 5% CO2 für 10 Minuten inkubiert. Für die EP3-Antagonismus-Messung wird der EP3-Rezeptor im FLIPR Tetra® durch Zugabe von 20 μΙ ~2 nM (2 x EC50, finale Konzentration) PGE2 in zum Beispiel Calcium-freier Tyrode / 2 mM CaC aktiviert und der Calcium-Flux durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt [Lit.: M. Kotani et al. (1995) Mol. Pharmacol. 48: 869-879; M. Kotani et al. (1997) Genomics 40: 425-434; T. Kunikata et al. (2005) Nat. Immunol. 6: 524-531 ; S. Narumiya und G. A. Fitzgerald (2001 ) J. Clin. Invest. 108: 25-30; F. Ushikubi et al. (1998) Nature 395: 281-284].
B-15. In vitro-Jest auf EP4-Agonismus und -Antagonismus
Für die Charakterisierung von Testsubstanzen auf EP4-Agonismus und -Antagonismus wurde der PGE2-induzierte Calcium-Flux in EP4-exprimierenden CHEM1 -Zellen (Millipore, HTS142C) verwendet: 3000 Zellen in 25 μΙ Plattiermedium [DMEM, 4.5 g/l Glucose, 4 mM Glutamin, 10%
Hitze-inaktiviertes FCS, 1 % 100x nicht-essentielle Aminosäuren, 10 mM HEPES, 100 U/ml Penicillin und Streptomycin] werden je Vertiefung einer 384-Multititerplatte (Fa. Greiner, TC-Platte, schwarz mit klarem Boden) ausgesät und bei 37°C / 5% CO2 für 24 Stunden inkubiert. Vor der Messung wird das Medium durch 30 μΙ Calcium-Farbstoff-Beladungspuffer (FLIPR Calcium As- say, Molecular Devices) ersetzt und bei 37°C / 5% CO2 für 60 Minuten inkubiert. Die Testsubstanz wird in DMSO in verschiedenen Konzentrationen als Dosiswirkungskurve vorbereitet (Startkonzentration 10 mM, Verdünnungsfaktor 3.16) und 1 :50 mit zum Beispiel Calcium-freier Tyrode (130 mM NaCI, 5 mM KCl, 20 mM HEPES, 1 mM MgCI2, 4.8 mM NaHCOs, pH 7.4) / 2 mM CaC vorverdünnt. Für die EP4-Agonismus-Messung werden in einem Fluoreszenzmess- gerät (FLIPR Tetra®, Molecular Devices) 10 μΙ der vorverdünnten Substanzlösung zu den mit Calcium-Farbstoff beladenen Zellen gegeben und der Calcium-Flux durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt. Danach werden die Zellen bei 37°C / 5% CO2 für 10 Minuten inkubiert. Für die EP4-Antagonismus-Messung wird der EP4- Rezeptor im FLIPR Tetra® durch Zugabe von 20 μΙ -26 nM (2 x EC50, finale Konzentration) PGE2 in zum Beispiel Calcium-freier Tyrode / 2 mM CaC aktiviert und der Calcium-Flux durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt [Lit. : S. Na- rumiya und G. A. Fitzgerald (2001 ) J. Clin. Invest. 108: 25-30; M. Nguyen et al. (1997) Nature 390: 78-81 ; K. Yoshida et al. (2002) Proc. Natl. Acad. Sei. USA 99: 4580-4585].
B-16. In vitro-Jest auf IP-Agonismus und -Antagonismus
Für die Charakterisierung von Testsubstanzen auf IP-Agonismus und -Antagonismus wurde der lloprost-induzierte Calcium-Flux in IP-exprimierenden CHEM1-Zellen (Millipore, HTS131 C) verwendet: 3000 Zellen in 25 μΙ Plattiermedium [DMEM, 4.5 g/l Glucose, 4 mM Glutamin, 10% Hit- ze-inaktiviertes FCS, 1 % 100x nicht-essentielle Aminosäuren, 10 mM HEPES, 100 U/ml Penicillin und Streptomycin] werden je Vertiefung einer 384-Multititerplatte (Fa. Greiner, TC-Platte, schwarz mit klarem Boden) ausgesät und bei 37°C / 5% CO2 für 24 Stunden inkubiert. Vor der Messung wird das Medium durch 30 μΙ Calcium-Farbstoff-Beladungspuffer (FLIPR Calcium As- say, Molecular Devices) ersetzt und bei 37°C / 5% CO2 für 60 Minuten inkubiert. Die Testsubstanz wird in DMSO in verschiedenen Konzentrationen als Dosiswirkungskurve vorbereitet (Startkonzentration 10 mM, Verdünnungsfaktor 3.16) und 1 :50 mit zum Beispiel Calcium-freier Tyrode (130 mM NaCI, 5 mM KCl, 20 mM HEPES, 1 mM MgCI2, 4.8 mM NaHCOs, pH 7.4) / 2 mM CaC vorverdünnt. Für die IP-Agonismus-Messung werden in einem Fluoreszenzmessgerät (FLIPR Tetra®, Molecular Devices) 10 μΙ der vorverdünnten Substanzlösung zu den mit Calcium-Farbstoff beladenen Zellen gegeben und der Calcium-Flux durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt. Danach werden die Zellen bei 37°C / 5% CO2 für 10 Minuten inkubiert. Für die IP-Antagonismus-Messung wird der IP-Rezeptor im FLIPR Tetra® durch Zugabe von 20 μΙ -106 nM (2 x EC50, finale Konzentration) lloprost in
zum Beispiel Calcium-freier Tyrode / 2 mM CaC aktiviert und der Calcium-Flux durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt [Lit. : S. Narumiya et al. (1999) P ysiol. Rev. 79: 1 193-1226; T. Murata et al. (1997) Nature 388: 678-682; Y. Cheng et al. (2002) Science 296: 539-541 ; C. H. Xiao et al. (2001 ) Circulation 104: 2210-2215; G. A. Fitz- gerald (2004) N. Engl. J. Med. 351 : 1709-171 1].
B-17. In vitro-Jest auf TP-Aqonismus und -Antagonismus
Für die Charakterisierung von Testsubstanzen auf TP-Agonismus und -Antagonismus wurde der 1146619-induzierte Calcium-Flux in TP-exprimierenden CHEM1 -Zellen (Millipore, HTS081 C) verwendet: 3000 Zellen in 25 μΙ Plattiermedium [DMEM, 10% Hitze-inaktiviertes FCS, 1 % 100x nicht-essentielle Aminosäuren, 10 mM HEPES, 0.25 mg/ml Geneticin (G418), 100 U/ml Penicillin und Streptomycin] werden je Vertiefung einer 384-Multititerplatte (Fa. Greiner, TC-Platte, schwarz mit klarem Boden) ausgesät und bei 37°C / 5% CO2 für 24 Stunden inkubiert. Vor der Messung wird das Medium durch 30 μΙ Calcium-Farbstoff-Beladungspuffer (FLIPR Calcium As- say, Molecular Devices) ersetzt und bei 37°C / 5% CO2 für 60 Minuten inkubiert. Die Testsub- stanz wird in DMSO in verschiedenen Konzentrationen als Dosiswirkungskurve vorbereitet (Startkonzentration 10 mM, Verdünnungsfaktor 3.16) und 1 :50 mit zum Beispiel Calcium-freier Tyrode (130 mM NaCI, 5 mM KCl, 20 mM HEPES, 1 mM MgCI2, 4.8 mM NaHCOs, pH 7.4) / 2 mM CaC vorverdünnt. Für die TP-Agonismus-Messung werden in einem Fluoreszenzmessgerät (FLIPR Tetra®, Molecular Devices) 10 μΙ der vorverdünnten Substanzlösung zu den mit Calcium-Farbstoff beladenen Zellen gegeben und der Calcium-Flux durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt. Danach werden die Zellen bei 37°C / 5% CO2 für 10 Minuten inkubiert. Für die TP-Antagonismus-Messung wird der TP- Rezeptor im FLIPR Tetra® durch Zugabe von 20 μΙ -88 nM (2 x EC50, finale Konzentration) U46619 in zum Beispiel Calcium-freier Tyrode / 2 mM CaC aktiviert und der Calcium-Flux durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt [Lit.: S. Ali et al. (1993) J. Biol. Chem. 268: 17397-17403; K. Hanasaki et al. (1989) Biochem. Phar- macol. 38: 2967-2976; M. Hirata et al. (1991 ) Nature 349: 617-620].
B-18. Tiermodell der Bleomycin-induzierten pulmonalen Fibrose
Die Bleomycin-induzierte Lungenfibrose bei der Maus oder Ratte ist ein weit verbreitetes Tier- modell für die Lungenfibrose. Bleomycin ist ein Glykopeptid-Antibiotikum, das in der Onkologie zur Therapie von Hodentumoren, Hodgkin- und Non-Hodgkin-Tumoren eingesetzt wird. Es wird renal eliminiert, besitzt eine Halbwertszeit von ca. 3 Stunden und beeinflusst als Zytostatikum verschiedene Phasen des Teilungszyklus [Lazo et al., Cancer Chemother. Biol. Response Modif. 15, 44-50 (1994)]. Sein anti-neoplastischer Effekt beruht auf einer oxidativ-schädigenden Wir- kung auf DNA [Hay et al., Arch. Toxicol. 65, 81-94 (1991 )]. Das Lungengewebe ist gegenüber Bleomycin in besonderer Weise gefährdet, da hier sog. Cysteinhydrolasen, welche in anderen
Geweben zu einer Inaktivierung von Bleomycin führen, nur in geringer Anzahl vorhanden sind. Nach Gabe von Bleomycin kommt es bei den Tieren zu einem "acute respiratory distress Syndrome" (ARDS) mit anschliessender Entwicklung einer Lungenfibrose.
Die Verabreichung des Bleomycins kann in einfacher oder mehrfacher Gabe intratracheal, inha- lativ, intravenös oder intraperitoneal erfolgen. Die Behandlung der Tiere mit der Testsubstanz (per gavage, durch Zusatz im Futter oder Trinkwasser, per osmotischer Minipumpe, per subkutaner oder intraperitonealer Injektion oder per Inhalation) beginnt am Tag der ersten Applikation des Bleomycins oder therapeutisch 3-14 Tage später und erstreckt sich über einen Zeitraum von 2-6 Wochen. Am Studienende werden eine bronchio-alveoläre Lavage zur Bestimmung des Zellgehaltes und der pro-inflammatorischen und pro-fibrotischen Marker sowie Messungen der Lungenfunktion und eine histologische Beurteilung der Lungenfibrose durchgeführt.
B-19. Tiermodell der DQ12-Quarz-induzierten pulmonalen Fibrose
DQ12-Quarz-induzierte Lungenfibrose an Maus und Ratte ist ein weit verbreitetes Tiermodell für Lungenfibrose [Shimbori et al., Exp. Lung Res. 36, 292-301 (2010)]. DQ12-Quarz ist ein durch Brechen beziehungsweise Mahlen hochaktiver Quarz. Die intratracheale oder inhalative Applikation von DQ12-Quarz führt bei Mäusen und Ratten zu einer Alveolarproteinose gefolgt von einer interstitiellen Lungenfibrose. Die Tiere erhalten eine einfache oder mehrfache intratracheale oder inhalative Instillation von DQ12-Quarz. Die Behandlung der Tiere mit der Testsubstanz (per gavage, durch Zusatz im Futter oder Trinkwasser, per osmotischer Minipumpe, per subkutaner o- der intraperitonealer Injektion oder per Inhalation) beginnt am Tag der ersten Instillation des Silikats oder therapeutisch 3-14 Tage später und erstreckt sich über einen Zeitraum von 3-12 Wochen. Am Studienende werden eine bronchio-alveoläre Lavage zur Bestimmung des Zellgehalts und der pro-inflammatorischen und pro-fibrotischen Marker sowie Messungen der Lungenfunktion und eine histologische Beurteilung der Lungenfibrose durchgeführt. B-20. Tiermodell der DQ12-Quarz-induzierten pulmonalen Inflammation
Eine intratracheale Gabe von DQ12-Quarz bei Maus und Ratte führt zu einer Inflammation in der Lunge [Shimbori et al., Exp. Lung Res. 36, 292-301 (2010)]. Die Tiere werden am Tag der Instillation von DQ12-Quarz oder einen Tag später für eine Dauer von 24 h bis zu 7 Tagen mit der Testsubstanz behandelt (per gavage, durch Zusatz im Futter oder Trinkwasser, per osmotischer Minipumpe, per subkutaner oder intraperitonealer Injektion oder per Inhalation). Am Versuchsende wird eine bronchio-alveoläre Lavage zur Bestimmung des Zellgehaltes und der proinflammatorischen und pro-fibrotischen Marker durchgeführt.
B-21. Tetrachlorkohlenstoff (CCU) induzierte Leberfibrose in Mäusen
Sechzig C57BL 6J Mäuse (Charles River, männlich, 8 Wochen alt) werden randomisiert und gleichermaßen in 4 Gruppen aufgeteilt (15 Mäuse pro Gruppe). Gruppe 1 dient als unbehandelte gesunde Kontrollgruppe, wohingegen die Gruppen 2-4 als an Leberfibrose erkrankte Mäuse die- nen. Die Leberfibrose wird durch intraperitoneale Injektion von 50 μΙ CCU-Olivenöl-Suspension (CCU + Olivenöl, 1 + 9 v/v) dreimal pro Woche (Montag, Mittwoch und Freitag) über den gesamten Studienzeitraum induziert. CCU ist die älteste und am meisten verwendete Substanz zur Auslösung toxisch-induzierter Leberfibrose (Starkel and Leclercq, Best Pract. Res. Clin. Gastro- enterol. 2011 , 25, 319-333). Die CCU behandelten Mäuse der Gruppe 2 dienen als Krankheits- kontrolle und erhalten keine weiteren Behandlungen, wohingegen die CCU behandelten Mäuse der Gruppe 3 zusätzlich mit Vehikel behandelt werden und damit als Vehikelkontrolle dienen. Die mit CCU behandelten Mäuse der Gruppe 4 dienen als mit einer Verbindung der Formel (I) behandelte Gruppe. Die orale Behandlung der Gruppe 3 mit der Vehikellösung und der Gruppe 4 mit einer Verbindung der Formel (I) startet am Tag 1 und wird zweimal täglich (morgens und abends) über den kompletten Studienzeitraum von zwei Wochen fortgeführt. Alle Tiere werden am Ende der Studie unter Betäubung getötet, die Lebern entnommen und in 4% gepufferte Formaldehydlösung für die nachfolgende histologische Aufarbeitung und Untersuchung fixiert. Dafür werden Leberproben aller Tiere in Paraffin eingebettet und 3 μηη dicke Paraffinschnitte hergestellt. Danach werden alle Leberschnitte deparaffiniert und mit Picro-Sirius-Rot (Waldeck, Ger- many) zur Bestimmung der Leberfibrose gefärbt. Die Picro-Sirius-Rot Färbung ist eine histologische Technik zur Färbung von Kollagen in Gewebe und damit von Fibrose. Ein Carl Zeiss Mikroskop (Axio Observer), welches mit einem Computer verbunden ist, wird zum Scannen der Pik- ro-Sirius-Rot gefärbten Leberschnitte zur Erzeugung von entsprechenden Bildern eingesetzt. Die Schnitte werden mit einer 20x Vergrößerung und einer Lichtintensität von 4,8 V gescannt. Die so erzeugten Bilder werden dann in JPG Format überführt und das rot gefärbte Areal mittels ImageJ Software (National Institute of Health, USA) quantifiziert. Die Ergebnisse werden in % Siriusrot pro Leberfläche dargestellt.
B-22. Tiermodell der unilateralen Ureterobstruktion (UUO) bei Mäusen (renalen Fibrose)
Die unilaterale Ureterobstruktion bei Maus und Ratte ist ein weit verbreitetes Tiermodell für in- terstitielle Nierenfibrose (Chevalier et al., Kidney Int. 2009, 75, 1145-1 152). Die dauerhafte Okklusion des Ureters führt durch die andauernde Stauung des Harns zur vermehrten inflammatorischen Zellinfiltration ins Interstitium, zu tubulären Zelltod und einem irreversiblen Verlust des Nierenparenchyms. Nach 5 bis 7 Tagen entsteht aufgrund der erhöhten Ablagerung von extrazellulären Matrixproteinen eine interstitielle Fibrose. Erwachsene männliche C57BI6J Mäuse (Charles River Labore, Sulzfeld, Deutschland) mit einem Gewicht von 20-22 g werden mit Isofluran betäubt, anschließend wird nach Eröffnung der Bauchhöhle ein Ureter ligiert und unterhalb der Li-
gatur durchtrennt. Bei den scheinoperierten Kontrollmäusen (SHAM) erfolgt nur die Eröffnung der Bauchhöhle, der Ureter wird aber nicht ligiert. Die Behandlung der Tiere in den Substanzgruppen wird nach dem Eingriff begonnen und für weitere 10 Tage fortgesetzt. 10 Tage nach der UUO werden die Tiere betäubt und durch Entbluten getötet. Danach werden die Nieren entnommen und die Beurteilung der renalen Fibrose erfolgt anhand der Expression von pro-inflammatorischen und pro-fibrotischen Markern sowie einer histologischen Beurteilung des Nierengewebes.
B-23. Silika induzierte Lungenfibrose in Mäusen: Therapeutische chronische 30 Tage Studie mit FP-Antagonisten
Erwachsene weibliche C57BI6J Mäuse (Charles River Labore, Sulzfeld, Deutschland) mit einem Gewicht von 18-20 g werden in einer Kammer mit Isofluran (3 % v/v) betäubt und intratracheal mit 2,5 mg kristallinem DQ12 Silika, welches in 70 μΙ steriler phosphatgepufferter Salzlösung gelöst war, behandelt. Unbehandelte Kontrollmäuse erhalten das gleiche Volumen mit phosphatgepufferter Salzlösung. Am Tag 10 nach der Silikabehandlung werden die Tiere in den Substanzgruppen für 20 Tage behandelt. 30 Tage nach der Silikainstillation werden die Tiere mit ei- ner intraperitonealen Injektion von Ketamin/Medetomidin (50 mg/kg und 0,33 mg/kg) kombiniert mit einer subkutanen Injektion von Temgesic (0,06 mg/kg) betäubt und durch Entbluten getötet. Danach wird die Trachea kanuliert und die Lungen der Tiere dreimal mit 0,5 ml eiskalter phosphatgepufferter Salzlösung lavagiert. Im Anschluss werden die Lungen entnommen, gewogen und auf Trockeneis schockgefroren. Nach Homogensation des Lungengewebes wird Hydroxy- prolin mittels HPLC bestimmt [Paroni et at., Clin. Chem. 1992, 38, 407-41 1 ; Säule: Phenomenex Synergi Hydro RP 4 μηη 80A, 75 χ 4,6 mm; Gradient: Laufmittel A: Wasser (6 ml/L Triethylamin, 3ml/L Essigsäure) pH 4,3; Lösemittel B: Acetonitril; Fluss: 1 ,3ml/min). Die Daten werden als Mittelwerte ± SEM von 8-12 Tieren pro Gruppe erhoben. Die statistische Analyse wird mit dem un- gepaarten Student's t-test durchgeführt. P-Werte mit < 0.05 werden als signifikant betrachtet. B-24. Effekte von FP-Antagonisten auf mechanische Sensitivität (periphär. Maus)
Mechanische Allodynie wurde unter Verwendung des von-Frey-Tests an den injizierten und nicht injizierten Hinterpfoten mehrere Male nach der Injektion untersucht.
Die mechanische Allodynie wurde mit 8 Semmes-Weinstein-Filamenten (Stölting ©; Wood Dale, IL, USA) mit unterschiedlicher Steifigkeit (0,04; 0,07; 0,16; 0,4; 1 ,0; 4,0 und 8,0 g) nach der Up- Down-Methode (Chaplan et al., J. Neurosci. Meth. 1994, 53, 56-63) gemessen. Intakte männliche ND4-Mäuse (~ 30 g,10 Tiere pro Gruppe) wurden in einzelnen Acrylkammern auf einer Metallgitteroberfläche platziert und konnten sich vor dem Testen für mindestens 15 Minuten an ihre Umgebung gewöhnen. Jedes Filament wurde senkrecht zur Pfotenunterseite mit ausreichender Kraft gedrückt, um ein leichtes Knicken gegen die Pfote zu verursachen, und wurde für ungefähr 6 Se- künden gehalten oder bis eine positive Antwort registriert wurde (Pfote scharf zurückgezogen).
Das Testen wurde mit dem 0,4 g-Filament begonnen. Wurde die Pfote nicht zurückgezogen, wurde der nächste stärkere Stimulus verwendet. Im Falle eines Zurückziehens der Pfote wurde der nächst schwächere Stimulus verwendet. Dieser Prozess wurde wiederholt, bis 4 Antworten nach der anfänglichen Änderung der Antwort (keine Antwort auf positive Antwort oder positive Antwort auf keine Antwort) erhalten werden. Reagierte das Tier nach Erreichen des stärksten Filaments nicht oder reagierte das Tier nach Erreichen des schwächsten Filaments, wird der Test für diesen Zeitpunkt abgebrochen. Die 50% -Antwortschwelle (50% Paw Response Threshold) wurde unter Verwendung der folgenden Formel berechnet:
10(X/+fcS)
50% Paw Response Threshold (g) =
10000
Xf = Wert (in logarithmischen Einheiten) des letzten verwendeten von Frey Filaments k = Tabellenwert für das Muster positiver / negativer Antworten (siehe Chaplan et al., J. Neuro- sci. Meth. 1994, 53, 56-63, Anhang 1 , Seite 62)
δ = mittlere Differenz (in logarithmischen Einheiten) zwischen den Stimuli
Der Mittelwert und Standardfehler des Mittelwerts (SEM) wurde für jede Pfote für jede Behandlungsgruppe zu jedem Zeitpunkt bestimmt.
Gruppendesign
IPL: intraplantar; PO: per os
Um den analgetischen Effekt eines FP-Antagonisten auf Fluprostenol-induzierte mechanische Sensitivität zu bewerten, wurde die Testsubstanz aus Bsp. 153 (90 mg/kg) 2 Stunden vor und 8 sowie 22 Stunden nach der Fluprostenol-Injektion verabreicht. Ipsilaterale und kontralaterale„50% Paw Response Thresholds" wurden vor der Verabreichung von Fluprostenol und bei 0,5, 2, 6 und
24 h nach der Verabreichung untersucht. In der mit 1 ,5 mg/kg Fluprostenol-behandelten Gruppe erhöhte die orale Verabreichung von Bsp. 153 (90 mg/kg) die„50% Paw Response Thresholds" 0,5 Stunden nach der Fluprostenol-Injektion im Vergleich zu Vehikel-behandelten Tieren signifikant. In der mit 0,15 mg/kg Fluprostenol-behandelten Gruppe erhöhte die orale Verabreichung von Bsp. 153 (90 mg/kg) die„50% Paw Response Thresholds" 24 Stunden nach der Fluprostenol- Injektion im Vergleich zu den mit Vehikel behandelten Tieren signifikant (siehe Tabelle 2).
Tabelle 2: 50% Paw Response Thresholds (g)
C. Ausführungsbeispiele für pharmazeutische Zusammensetzungen Die erfindungsgemäßen Verbindungen können folgendermaßen in pharmazeutische Zubereitungen überführt werden:
Tablette:
Zusammensetzung:
100 mg der erfindungsgemäßen Verbindung, 50 mg Lactose (Monohydrat), 50 mg Maisstärke (nativ), 10 mg Polyvinylpyrrolidon (PVP 25) (Fa. BASF, Ludwigshafen, Deutschland) und 2 mg Magnesiumstearat.
Tablettengewicht 212 mg. Durchmesser 8 mm, Wölbungsradius 12 mm.
Herstellung:
Die Mischung aus erfindungsgemäßer Verbindung, Lactose und Stärke wird mit einer 5%-igen Lösung (m/m) des PVPs in Wasser granuliert. Das Granulat wird nach dem Trocknen mit dem Magnesiumstearat 5 Minuten gemischt. Diese Mischung wird mit einer üblichen Tablettenpresse verpresst (Format der Tablette siehe oben). Als Richtwert für die Verpressung wird eine Presskraft von 15 kN verwendet.
Oral applizierbare Suspension:
Zusammensetzunq:
1000 mg der erfindungsgemäßen Verbindung, 1000 mg Ethanol (96%), 400 mg Rhodigel® (Xan- than gum der Firma FMC, Pennsylvania, USA) und 99 g Wasser.
Einer Einzeldosis von 100 mg der erfindungsgemäßen Verbindung entsprechen 10 ml orale Sus- pension.
Herstellung:
Das Rhodigel wird in Ethanol suspendiert, die erfindungsgemäße Verbindung wird der Suspension zugefügt. Unter Rühren erfolgt die Zugabe des Wassers. Bis zum Abschluß der Quellung des Rhodigels wird ca. 6 h gerührt. Oral applizierbare Lösung:
Zusammensetzung:
500 mg der erfindungsgemäßen Verbindung, 2.5 g Polysorbat und 97 g Polyethylenglycol 400. Einer Einzeldosis von 100 mg der erfindungsgemäßen Verbindung entsprechen 20 g orale Lösung.
Herstellung:
Die erfindungsgemäße Verbindung wird in der Mischung aus Polyethylenglycol und Polysorbat unter Rühren suspendiert. Der Rührvorgang wird bis zur vollständigen Auflösung der erfindungsgemäßen Verbindung fortgesetzt. i.v. -Lösung:
Die erfindungsgemäße Verbindung wird in einer Konzentration unterhalb der Sättigungslöslich- keit in einem physiologisch verträglichen Lösungsmittel (z.B. isotonische Kochsalzlösung, Glu- coselösung 5% und/oder PEG 400-Lösung 30%) gelöst. Die Lösung wird steril filtriert und in sterile und pyrogenfreie Injektionsbehältnisse abgefüllt.